Protein Purification Methods
For ordering information on the products discussed here, please visit our Protein Purification product pages.
Introduction to Protein Purification
A fundamental step in studying individual proteins is purification of the protein of interest. There are four basic steps of protein purification: 1) cell lysis, 2) protein binding to a matrix, 3) washing and 4) elution. Cell lysis can be accomplished a number of ways, including nonenzymatic methods (e.g., sonication or French press) or use of hydrolytic enzymes such as lysozyme or a detergent reagent such as FastBreak™ Cell Lysis Reagent. Because purification of native proteins can be challenging, affinity purification tags are often fused to a recombinant protein of interest such that the tag is used to capture or detect the protein.
Here we provide an overview of protein purification strategies, including guidelines on choosing a purification method and example protocols for protein purification using affinity tags.
Protein Purification Strategies
Proteins are biological macromolecules that maintain the structural and functional integrity of the cell, and many diseases are associated with protein malfunction. Protein purification is a fundamental step for analyzing individual proteins and protein complexes and identifying interactions with other proteins, DNA or RNA. A variety of protein purification strategies exist to address desired scale, throughput and downstream applications. The optimal approach often must be determined empirically.
Protein Purification
The best protein purification protocol depends not only on the protein being purified but also on many other factors such as the cell used to express the recombinant protein (e.g., prokaryotic versus eukaryotic cells). Escherichia coli remains the first choice of many researchers for producing recombinant proteins due to ease of use, rapid cell growth and low cost of culturing. Proteins expressed in E. coli can be purified in relatively high quantities, but these proteins, especially eukaryotic proteins, may not exhibit proper protein activity or folding. Cultured mammalian cells might offer a better option for producing properly folded and functional mammalian proteins with appropriate post-translational modifications (Geisse et al. 1996). However, the low expression levels of recombinant proteins in cultured mammalian cells presents a challenge for their purification. As a result, attaining satisfactory yield and purity depends on highly selective and efficient capture of these proteins from the crude cell lysates.
To simplify purification, affinity purification tags can be fused to a recombinant protein of interest (Nilsson et al. 1997). Common fusion tags are polypeptides, small proteins or enzymes added to the N or C terminus of a recombinant protein. The biochemical features of different tags influence the stability, solubility and expression of proteins to which they are attached (Stevens et al. 2001). Using expression vectors that include a fusion tag facilitates recombinant protein purification.
Isolation of Protein Complexes
A major objective in proteomics is the elucidation of protein function and organization of the complex networks that are responsible for key cellular processes. Analysis of protein:protein interactions can provide valuable insight into the cell signaling cascades involved in these processes, and analysis of protein:nucleic acid interactions often reveals important information about biological processes such as mRNA regulation, chromosomal remodeling and transcription. For example, transcription factors play an important role in regulating transcription by binding to specific recognition sites on the chromosome, often at a gene’s promoter, and interacting with other proteins in the nucleus. This regulation is required for cell viability, differentiation and growth (Mankan et al. 2009; Gosh et al. 1998).
Analysis of protein:protein interactions often requires straightforward methods for immobilizing proteins on solid surfaces in proper orientations without disrupting protein structure or function. This immobilization must not interfere with the binding capacity and can be achieved through the use of affinity tags. Immobilization of proteins on chips is a popular approach to analyze protein:DNA and protein:protein interactions and identify components of protein complexes (Hall et al. 2004; Hall et al. 2007; Hudson and Snyder, 2006). Functional protein microarrays normally contain full-length functional proteins or protein domains bound to a solid surface. Fluorescently labeled DNA is used to probe the array and identify proteins that bind to the specific probe. Protein microarrays provide a method for high-throughput identification of protein:DNA interactions. Immobilized proteins also can be used in protein pull-down assays to isolate protein binding partners in vivo (mammalian cells) or in vitro. Other downstream applications such as mass spectrometry do not require protein immobilization to identify protein partners and individual components of protein complexes.
Affinity Tags for Protein Purification
One method for isolating or immobilizing a specific protein is the use of affinity tags. Many different affinity tags have been developed (Terpe, 2002). Fusion tags are polypeptides, small proteins or enzymes added to the N or C terminus of a recombinant protein.
Polyhistidine
The most commonly used tag is the polyhistidine tag (Yip et al. 1989). Protein purification using the polyhistidine tag relies on the affinity of histidine residues for immobilized metal such as nickel (Yip et al. 1989; Hutchens and Yip, 1990). This affinity interaction is believed to be a result of coordination of a nitrogen on the imidazole moiety of polyhistidine with a vacant coordination site on the metal. The metal is immobilized to a support through complex formation with a chelate that is covalently attached to the support.
Polyhistidine tags offer several advantages for protein purification. The small size of the polyhistidine tag renders it less immunogenic than other larger tags. Therefore, the tag usually does not need to be removed for downstream applications following purification. A polyhistidine tag may be placed on either the N- or C-terminus of the protein of interest. Finally, the interaction of the polyhistidine tag with the metal does not depend on the tertiary structure of the tag, making it possible to purify otherwise insoluble proteins using denaturing conditions.
Glutathione-S-Transferase
The use of the affinity tag glutathione-S-transferase (GST) is based on the strong affinity of GST for immobilized glutathione-covered matrices (Smith and Johnson, 1988). Glutathione-S-transferases are a family of multifunctional cytosolic proteins that are present in eukaryotic organisms (Mannervik and Danielson, 1988; Armstrong, 1997). GST isoforms are not normally found in bacteria; thus endogenous bacterial proteins don’t compete with the GST-fusion proteins for binding to the purification resin. The 26kDa GST affinity tag enhances the solubility of many eukaryotic proteins expressed in bacteria.
HaloTag® Protein Tag
Protein fusion tags are used to aid expression of suitable levels of soluble protein as well as purification. A unique protein tag, the HaloTag® protein, is engineered to enhance expression and solubility of recombinant proteins in E. coli. HaloTag® protein tag is a 34kDa, monomeric protein tag modified from Rhodococcus rhodochrous dehalogenase.
The HaloTag® protein is designed to bind rapidly and covalently to a unique synthetic linker to achieve an irreversible attachment. The synthetic linker can be attached to a variety of entities such as fluorescent dyes and solid supports to allow labeling of fusion proteins in cell lysates for expression screening and capture of fusion proteins on a purification resin.
HaloTag® Technology is compatible with many protein expression systems and can be applied to proteins expressed in E. coli, mammalian cells and cell-free systems.
HaloTag® Technology
HaloTag is a powerful technology with applications for protein purification, protein localization, trafficking and turnover as well as protein interactions and super-resolution microscopy.
The lack of an endogenous equivalent of the HaloTag® protein in mammalian cells minimizes the chances of detecting false positives or nonspecific interactions. The combination of covalent capture and rapid binding kinetics overcomes the equilibrium-based limitations associated with traditional affinity tags and enables efficient capture even at low expression levels. In addition, the highly stable HaloTag® protein:ligand interaction permits boiling the protein complex in SDS sample buffer prior to SDS-PAGE analysis.
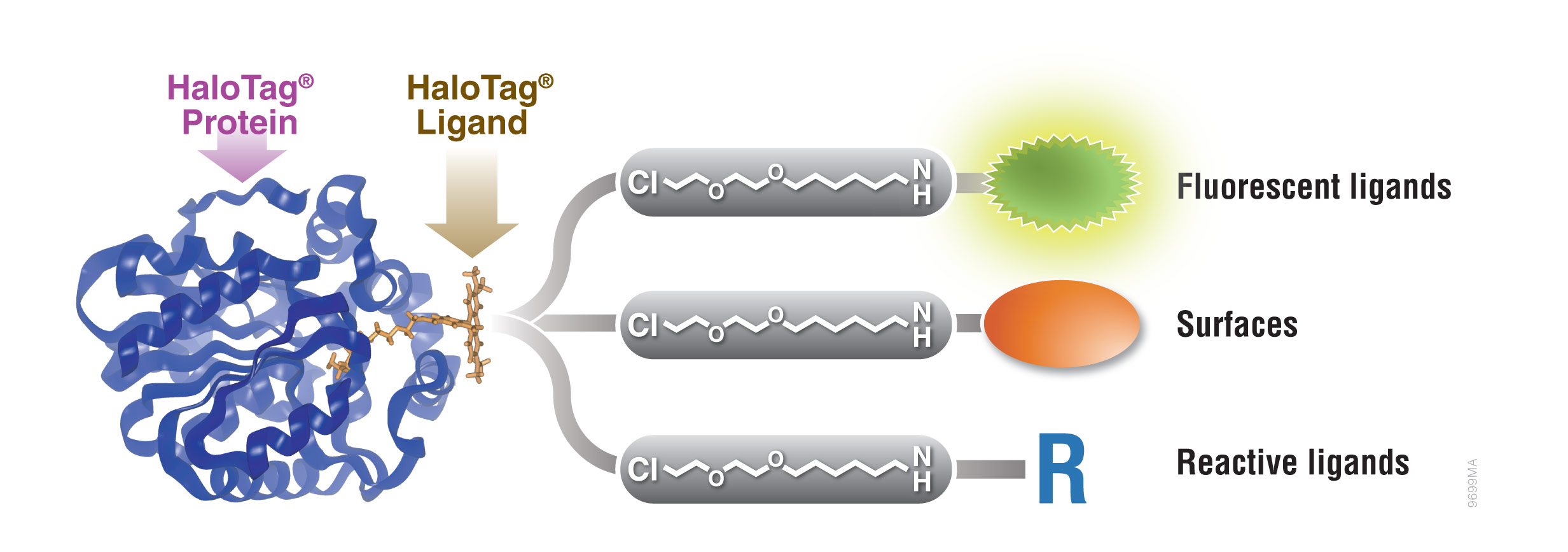
Figure 1. Interchangeable functionality of the HaloTag® protein tag. A covalent bond forms between the HaloTag® fusion protein and HaloTag® Ligand’s reactive linker under general physiological conditions. This interaction is highly specific and irreversible. Different HaloTag® Ligands with different functionalities are available to eliminate the need to design and create a new expression construct.
Purification of Polyhistidine-Tagged Proteins
Rapid Purification of Polyhistidine-Tagged Proteins Using Magnetic Resins
There is a growing need for high-throughput protein purification methods. Magnetic resins enable affinity-tagged protein purification without the need for multiple centrifugation steps and sequential transfer of samples to multiple tubes. There are several criteria that define a good protein purification resin: minimal nonspecific protein binding, high binding capacity for the fusion protein and efficient recovery of the fusion protein. The MagneHis™ Protein Purification System meets these criteria, enabling purification of proteins with a broad range of molecular weights and different expression levels. The magnetic nature of the binding particles allows purification from crude lysates to be performed in a single tube. In addition, the system can be used with automated liquid-handling platforms for high-throughput applications.
MagneHis™ Protein Purification System
The MagneHis™ Protein Purification System uses paramagnetic precharged nickel particles (MagneHis™ Ni-Particles) to isolate polyhistidine-tagged protein directly from a crude cell lysate. Figure 2 shows a schematic diagram of the MagneHis™ Protein Purification System protocol. Polyhistidine-tagged protein can be purified on a small scale using less than 1ml of culture or on a large scale using more than 1 liter of culture. Samples can be processed in a high-throughput manner using a robotic platform such as the Beckman Coulter Biomek® FX or Tecan Freedom EVO® instrument. Polyhistidine-tagged proteins can be purified under native or denaturing (2–8M urea or guanidine-HCl) conditions. The presence of serum in mammalian and insect cell culture medium does not interfere with purification. For more information and a detailed protocol, see Technical Manual #TM060 and the MagneHis™ Protein Purification System Automated Protocol #EP011.
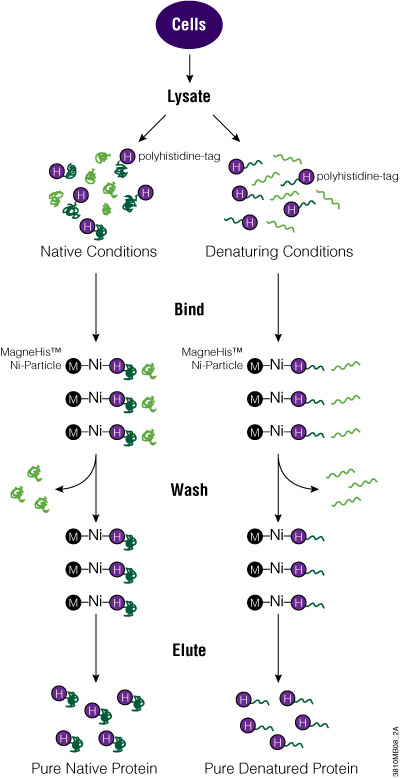
Figure 2. Diagram of the MagneHis™ Protein Purification System protocol.
Protocol: MagneHis™ Purification of Proteins Expressed in Bacterial Cells
Materials Required:
- MagneHis™ Protein Purification System and protocol
- 37°C incubator for flasks or tubes
- shaker
- magnetic separation stand
- 1M imidazole solution (pH 8.0; for purification from insect or mammalian cells or culture medium)
- additional binding/wash buffer (may be required if processing numerous insect cell, mammalian cell or culture medium samples)
- solid NaCl (for purification from insect or mammalian cells or culture medium)
Purification using Denaturing Conditions. Proteins expressed in bacterial cells may be present in insoluble inclusion bodies. To determine if your protein is located in an inclusion body, perform the lysis step using FastBreak™ Cell Lysis Reagent, 10X, as described in Technical Manual #TM060. Pellet cellular debris by centrifugation, and check the supernatant and pellet for the polyhistidine-tagged protein by gel analysis.
Efficient purification of insoluble proteins requires denaturing conditions. Since the interaction of polyhistidine-tagged fusion proteins and MagneHis™ Ni-Particles does not depend on tertiary structure, fusion proteins can be captured and purified using denaturing conditions by adding a strong denaturant such as 2–8M guanidine hydrochloride or urea to the cells. Denaturing conditions must be used throughout the procedure so that the proteins do not aggregate. We recommend preparing denaturing buffers by adding solid guanidine-HCl or urea directly to the MagneHis™ Binding/Wash and Elution Buffers. For more information, see Technical Manual #TM060.
Note: Do not combine FastBreak™ Cell Lysis Reagent and denaturants. Cells can be lysed directly using denaturants such as urea or guanidine-HCl.
Purification from Insect and Mammalian Cells. Process cells at a cell density of 2 × 106 cells/ml of culture. Adherent cells may be removed from the tissue culture vessel by scraping and resuspending in culture medium to this density. Cells may be processed in culture medium containing up to 10% serum. Processing more than the indicated number of cells per milliliter of sample may result in reduced protein yield and increased nonspecific binding. For proteins that are secreted into the cell culture medium, remove any cells from the medium prior to purification. For more information, see Technical Manual #TM060.
MagneHis™ System Protocols
More information and detailed protocols for use of the MagneHis™ System are available in Technical Manual #TM060. A protocol for automating MagneHis™ system on liquid handlers is also available (#EP011).
Medium- to Large-Scale Purification of Polyhistidine-Tagged Proteins In Column or Batch Formats
The two most common support materials for resin-based, affinity-tagged protein purification are agarose and silica gel. As a chromatographic support, silica is advantageous because it has a rigid mechanical structure that is not vulnerable to swelling and can withstand large changes in pressure and flow rate without disintegrating or deforming. Silica is available in a wide range of pore and particle sizes including macroporous silica, which provides a higher capacity for large biomolecules such as proteins. However, two of the drawbacks of silica as a solid support for affinity purification are the limited reagent chemistry that is available and the relatively low efficiency of surface modification.
The HisLink™ Protein Purification Resin (Cat.# V8821) overcomes these limitations by using a new modification process for silica surfaces that provides a tetradentate metal-chelated solid support with a high binding capacity and concomitantly eliminates the nonspecific binding that is characteristic of unmodified silica. HisLink™ Resin is a macroporous silica resin modified to contain a high level of tetradentate-chelated nickel (>20mmol Ni/ml settled resin). Figure 3 shows a schematic diagram of HisLink™ Resin and polyhistidine tag interaction. The HisLink™ Resin has a pore size that results in binding capacities as high as 35mg of polyhistidine-tagged protein per milliliter of resin.
The HisLink™ Resin enables efficient capture and purification of bacterially expressed polyhistidine-tagged proteins. This resin also may be used for general applications that require an immobilized metal affinity chromatography (IMAC) matrix (Porath et al. 1975; Lonnerdal and Keen, 1982). HisLink™ Resin may be used in either column or batch purification formats. For a detailed protocol, see Technical Bulletin #TB327.
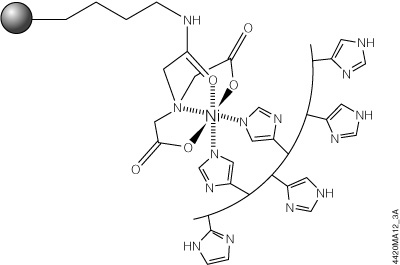
Figure 3. Schematic diagram of HisLink™ Resin and polyhistidine interaction. Two sites are available for polyhistidine-tag binding and are rapidly coordinated with histidine in the presence of a polyhistidine-tagged polypeptide.
Column-Based Purification using HisLink™ Resin
The HisLink™ Resin provides a conventional means to purify polyhistidine-tagged proteins and requires only a column that can be packed to the appropriate bed volume. When packed to 1ml under gravity-driven flow, HisLink™ Resin shows an average flow rate of approximately 1ml/minute. In general a flow rate of 1–2ml/minute per milliliter of resin is optimal for efficient capture of polyhistidine-tagged protein. Gravity flow of a cleared lysate over a HisLink™ column will result in complete capture and efficient elution of polyhistidine-tagged proteins; however, the resin also may be used with vacuum filtration devices (e.g., Vac-Man® Vacuum Manifold, Cat.# A7231) to allow simultaneous processing of multiple columns. HisLink™ Resin is also an excellent choice for affinity purification using low- to medium-pressure liquid chromatography systems such as fast performance liquid chromatography (FPLC).
Example Protocol Using the HisLink™ Resin to Purify Proteins from Cleared Lysate by Gravity-Flow Column Chromatography
Materials Required:
- HisLink™ Protein Purification Resin (Cat.# V8821) and protocol
- HEPES buffer (pH 7.5)
- imidazole
- HisLink™ Binding Buffer
- HisLink™ Wash Buffer
- HisLink™ Elution Buffer
- column
Cell Lysis: Cells may be lysed using any number of methods including sonication, French press, bead milling, treatment with lytic enzymes (e.g., lysozyme) or use of a commercially available cell lysis reagent such as the FastBreak™ Cell Lysis Reagent (Cat.# V8571). If lysozyme is used to prepare a lysate, add salt (>300mM NaCl) to the binding and wash buffers to prevent lysozyme binding to the resin. Adding protease inhibitors such as 1mM PMSF to cell lysates does not inhibit binding or elution of polyhistidine-tagged proteins with the HisLink™ Resin and is highly recommended to prevent degradation of the protein of interest by endogenous proteases. When preparing cell lysates from high-density cultures, adding DNase and RNase (concentrations up to 20μg/ml) will reduce the lysate viscosity and aid purification.
- Prepare the HisLink™ Binding, Wash and Elution Buffers
Note: Polyhistidine-tagged proteins can be eluted using 250–1,000mM imidazole. Polyhistidine tags containing less than six histidines typically require less imidazole for elution, while polyhistidine proteins containing more than six polyhistidines may require higher levels of imidazole. - Determine the column volume required to purify the protein of interest. In most cases 1ml of settled resin is sufficient to purify the amount of protein typically found in up to 1 liter of culture (cell density of O.D.600 < 6.0). In cases of very high expression levels (e.g., 50mg protein/liter), up to 2ml of resin per liter of culture may be needed.
- Once you have determined the volume of settled resin required, precalibrate this amount directly in the column by pipetting the equivalent volume of water into the column and marking the column to indicate the top of the water. This mark indicates the top of the settled resin bed. Remove the water before adding resin to the column.
- Make sure that the resin is fully suspended; fill the column with resin to the line marked on the column by transferring the resin with a pipette. Allow the resin to settle, and adjust the level of the resin by adding or removing resin as necessary.
Note: If the resin is not pipetted within 10–15 seconds of mixing, significant settling will occur, and the resin will need to be resuspended. Alternatively, a magnetic stir bar may be used to keep the resin in suspension during transfer. To avoid fracturing the resin, do not leave the resin stirring any longer than the time required to pipet and transfer the resin. - Allow the column to drain, and equilibrate the resin with five column volumes of binding buffer, allowing the buffer to completely enter the resin bed.
- Gently add the cleared lysate to the resin until the lysate has completely entered the column. The rate of flow through the column should not exceed 1–2ml/minute for every 1ml of column volume. Under normal gravity flow conditions the rate is typically about 1ml/minute. The actual flow rate will depend on the type of column used and the extent to which the lysate was cleared and filtered. Do not let the resin dry out after you have applied the lysate to the column.
- Wash unbound proteins from the resin using at least 10–20 column volumes of wash buffer. Divide the total volume of wash buffer into two or three aliquots, and allow each aliquot to completely enter the resin bed before adding the next aliquot.
- Once the wash buffer has completely entered the resin bed, add elution buffer and begin collecting fractions (0.5–5ml fractions). Elution profiles are protein-dependent, but polyhistidine-tagged proteins will generally elute in the first 1ml. Elution is usually complete after 3–5ml of buffer is collected per 1.0ml of settled resin, provided the imidazole concentration is high enough to efficiently elute the protein of interest.
Batch Protein Purification Using HisLink™ Resin
One of the primary advantages of the HisLink™ Resin is its use in batch purification. In batch mode, the protein of interest is bound to the resin by mixing lysate with the resin for approximately 30 minutes at a temperature range of 4–22°C. Once bound with protein, the resin is allowed to settle to the bottom of the container, and the spent lysate is removed. Washing requires only resuspension of the resin in an appropriate wash buffer followed by a brief period to allow the resin to settle. The wash buffer is then carefully poured off. This process is repeated as many times as desired. Final elution is best achieved by transferring the HisLink™ Resin to a column to elute the protein in fractions. The advantages of batch purification are: 1) less time is required to perform the purification; 2) large amounts of lysate can be processed; and 3) clearing the lysate prior to purification is not required.
Purification of Polyhistidine-Tagged Proteins by FPLC
The rigid particle structure of the silica base used in the HisLink™ Resin make this material an excellent choice for applications that require applied pressure to load the lysate, wash or elute protein from the resin. These applications involve both manual and automated systems that operate under positive or negative pressure (e.g., FPLC and vacuum systems, respectively). To demonstrate the use of HisLink™ Resin on an automated platform we used an AKTA explorer from GE Healthcare to purify milligram quantities of polyhistidine-tagged protein from 1 liter of culture. The culture was lysed in 20ml of binding/wash buffer and loaded onto a column containing 1ml of HisLink™ Resin. We estimate the total amount of protein recovered to be 75–90% of the protein expressed in the original lysate.
Protein purification under denaturing conditions: Proteins that are expressed as an inclusion body and have been solubilized with chaotrophic agents such as guanidine-HCl or urea can be purified by modifying the protocol to include the appropriate amount of denaturant (up to 6M guanidine-HCl or up to 8M urea) in the binding, wash and elution buffers.
HisLink™ Resin may be used in either column or batch purification formats. For a detailed protocol, see Technical Bulletin #TB327.
Purification of HaloTag® Fusion Proteins
HaloTag® Protein Purification from Mammalian Cells
Cultured mammalian cells offer an environment well suited for producing properly folded and functional mammalian proteins with appropriate post-translational modifications. However, the low expression levels of recombinant proteins in cultured mammalian cells presents a challenge. As a result, attaining satisfactory yield and purity depends on selective and efficient capture of these proteins from the crude cell lysate. The equilibrium-based binding of most affinity tag protein purification methods means that the protein is constantly being exchanged between the bound (to the resin) and unbound state. This equilibrium depends on the protein concentration and binding affinity of the tag. As a result, binding efficiency may be reduced at low expression levels, leading to low recovery of the fusion protein.
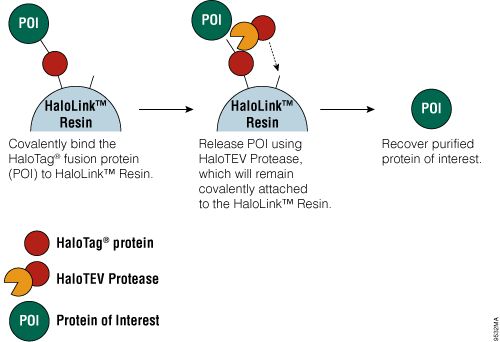
Figure 4. Schematic diagram of protein purification using HaloTag® Technology.
The HaloTag® Mammalian Protein Purification Systems (Cat.# G6795 and Cat.# G6790) use the HaloTag® protein tag, which can be genetically fused to any protein and transiently or stably expressed in mammalian cells. Following cell lysis, the HaloTag® fusion protein is covalently captured on the HaloLink™ Resin, and nonspecific proteins are washed away. The protein of interest is released by a specific proteolytic cleavage at an optimized TEV recognition site contained within the amino acid linker sequence that connects the HaloTag® protein tag and protein of interest. To eliminate the need for a secondary step to remove the protease, TEV protease fused to HaloTag® (HaloTEV Protease; Cat.# G6601) can be used to cleave the HaloTag® fusion protein and then covalently captured on the HaloLink™ Resin, resulting in a streamlined purification process. This straightforward purification uses a single, mild physiological buffer throughout the entire process with no need for buffer exchange.
HaloTag® Protein Purification from E. coli
The HaloTag® Protein Purification System (Cat.# G6280) allows covalent, efficient and specific capture of proteins expressed in E. coli as N-terminal HaloTag® fusion proteins. Many of the same characteristics that make the HaloTag® protein well suited for purifying proteins from mammalian cells also make it a good choice for purifying proteins from E. coli cells. HaloTag® fusion proteins can be expressed in E. coli using a number of expression vectors specifically designed for E. coli including the pFN18A HaloTag® T7 Flexi® Vector (Cat.# G2751) and pFN18K HaloTag® T7 Flexi® Vector (Cat.# G2681) as well as non-Flexi® vectors, which are available with dual tags of HaloTag® protein and polyhistidine. These non-Flexi® vectors, pH6HTN His6HaloTag® T7 Vector (Cat.# G7971) and pH6HTC His6HaloTag® T7 Vector (Cat.# G8031), allow traditional cloning using the multiple cloning site. These dual-tagged vectors enable purification of HaloTag® fusion proteins that still retain the covalent coupling ability of the HaloTag® protein. With the HaloTag® Protein Purification System, it is easy to perform in-gel detection and quantification of protein expression levels using fluorescent HaloTag® Ligands.
HaloTag® Protein Purification System Protocols
For detailed protocols on use of the HaloTag® Mammalian Protein Detection and Purification System, see Technical Manual, TM348. For protocols covering HaloTag® protein purification from E. coli, see the HaloTag® Protein Purification System Technical Manual, TM312.
Purification of Biotinylated Proteins
SoftLink™ Soft Release Avidin Resin
Biotinylated fusion proteins can be affinity-purified using the SoftLink™ Soft Release Avidin Resin. This proprietary resin allows elution of a fusion protein under native conditions by adding exogenous biotin.
Avidin:biotin interactions are so strong that elution of biotin-tagged proteins from avidin-conjugated resins usually requires denaturing conditions. In contrast, the SoftLink™ Soft Release Avidin Resin, which uses monomeric avidin, allows the protein to be eluted with a nondenaturing 5mM biotin solution. The rate of dissociation of the monomeric avidin-biotin complex is sufficiently fast to effectively allow recovery of all bound protein in neutral pH and low salt conditions.
The SoftLink™ Soft Release Avidin Resin is highly resistant to many chemical reagents (e.g., 0.1N NaOH, 50mM acetic acid and nonionic detergents), permitting stringent wash conditions.
SoftLink™ Soft Release Avidin Resin Protocol
A protocol for use of the SoftLink™ Soft Release Avidin Resin is available in Product Information Sheet 9PIV201.
Pull-Down Methods for Purifying Protein Complexes
HaloTag® Pull-Down Assays
Traditional protein pull-down approaches rely on binding of a protein to an affinity resin, and often this is not a very efficient process. The HaloTag® Mammalian Pull-Down Systems also rely on binding of the protein of interest to an affinity resin, but the HaloTag® protein fusion tag binds to the resin rapidly, covalently and irreversibly, unlike many other tags. These properties increase the chance of capturing protein complexes and retaining them after capture. In addition, the lack of an endogenous equivalent of the HaloTag® protein in mammalian cells minimizes the chance of detecting false positives or nonspecific interactions. An overview of the HaloTag® Mammalian Protein Pull-Down System protocol is depicted in Figure 5. More information and detailed protocols for the HaloTag® Mammalian Pull-Down and Labeling System (Cat.# G6500) and HaloTag® Mammalian Pull-Down System (Cat.# G6504) are available in Technical Manual #TM342. Information about the HaloTag® Complete Pull-Down System (Cat.# G6509) is available in Technical Manual #TM360.
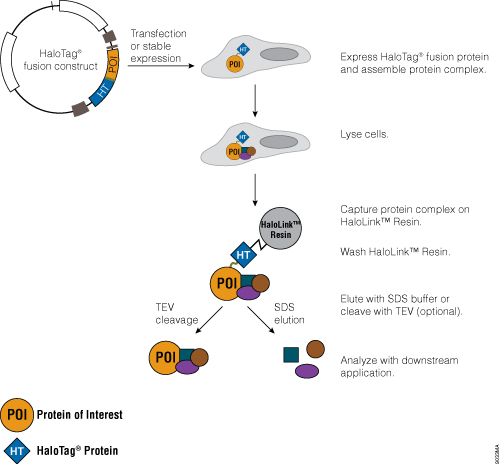
Figure 5. Schematic overview of the HaloTag® Mammalian Pull-Down System protocol.
HaloTag® Pull-Down Assay Protocols
See the HaloTag® Mammalian Pull-Down and Labeling Systems Technical Manual, TM342, and the HaloTag® Complete Pull-Down System Technical Manual, TM360, for detailed protocols for use of HaloTag® Pull-Down Assays.
References
- Armstrong, R.N. (1997) Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem. Res. Toxicol. 10, 2–18.
- Geisse, S. et al. (1996) Eukaryotic expression systems: A comparison. Protein Expr. Purif. 8, 271–82.
- Gosh, S. et al. (1998) NFκB and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 16, 225–60.
- Hall, D.A. et al. (2007) Protein microarray technology. Mech. Ageing Dev. 128, 161–7.
- Hall, D.A. et al. (2004) Regulation of gene expression by a metabolic enzyme. Science 306, 482–4.
- Hudson, M.E. and Snyder, M. (2006) High-throughput methods of regulatory element discovery. Biotechniques 41, 673–81.
- Hutchens, T.W. and Yip, T.T. (1990) Differential interaction of peptides and protein surface structures with free metal ions and surface-immobilized metal ions. J. Chromatogr. 500, 531–42.
- Lonnerdal, B. and Keen, C. (1982) Metal chelate affinity chromatography of proteins J. Appl. Biochem. 4, 203–8.
- Mankan, A.K. et al. (2009) NF-kappaB regulation: The nuclear response. J. Cell. Mol. Med. 13, 631–43.
- Mannervik, B. and Danielson, U.H. (1988) Glutathione transferases—structure and catalytic activity. CRC Crit. Rev. Biochem. 23, 283–337.
- Nilsson, J. et al. (1997) Affinity fusion strategies for detection, purification, and immobilization of recombinant proteins. Protein Expr. Purif. 11, 1–16.
- Porath, J. et al. (1975) Metal chelate affinity chromatography, a new approach to protein fractionation. Nature 258, 598–9.
- Smith, D.B. and Johnson, K.S. (1988) Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67, 31–40.
- Stevens, R.C. et al. (2001) Global efforts in structural genomics. Science 294, 89–92.
- Terpe, K. (2002) Overview of tag protein fusions: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 60, 523–33.
- Yip, T.T. et al. (1989) Evaluation of the interaction of peptides with Cu(II), Ni(II), and Zn(II) by high-performance immobilized metal ion affinity chromatography. Anal. Biochem. 183, 159–71.