Reporter Genes and their Applications
This guide provides an overview of bioluminescent reporter genes, including guidance for choosing a reporter and choosing an appropriate vector. Visit the Reporter Gene Vectors product page to view ordering information for the Reporter Vectors and Cell Lines discussed here.
Introduction to Reporter Genes
What is a Reporter Gene?
A reporter gene is an exogenous coding region joined to a promoter sequence or element in an expression vector that is introduced into cells to provide the means for measuring the promoter activity. Once expressed in cells, the reporter is assayed by either directly measuring the reporter protein itself or assessing its enzymatic activity, correlating the strength of the fused promoter element with the amount of reporter produced. The ideal reporter gene is not endogenously expressed in the cell of interest and amenable to assays that are sensitive, quantitative, rapid, easy, accurate, reproducible and safe.
What is the Role of a Reporter Gene?
Reporter genes and their associated detection methods have contributed greatly to the study of eukaryotic gene expression and regulation. Although reporter genes have played a significant role in numerous applications, both in vitro and in vivo, they are most frequently used as indicators of transcriptional activity in cells.
Analyzing cis-acting transcriptional elements is one common use of reporter genes. Reporter vectors, plasmids that contain the reporter gene, are used to identify and characterize promoter and enhancer element function because reporter protein expression corresponds with transcriptional activity of the reporter gene. For these types of studies, promoter sequences are cloned upstream or downstream of the gene. The promoter-reporter gene fusion is introduced into cultured cells by standard transfection methods or into a germ cell to produce transgenic organisms. Thus, you can characterize promoter and enhancer elements that regulate cell-, tissue- and development-defined gene expression by using reporter genes.
To assay trans-acting factors, the promoter-reporter gene fusion DNA is cotransfected with another cloned DNA expressing a trans-acting protein or RNA of interest or by activating the trans-acting factors through sample treatment. The protein could be a transcription factor that binds to the promoter region of interest cloned upstream of the reporter gene. For example, when tat protein, a transactivating protein in HIV-1, is expressed from one vector in a transfected cell, the activity of different HIV-1 long-terminal repeat (LTR) sequences linked to a reporter gene increases, shown by a corresponding increase of reporter gene protein activity.
In addition to assessing promoter activity, reporters are commonly used to study the effect of untranslated regions (UTRs) on mRNA stability, protein localization and translation efficiency. For example, microRNA (miRNA) target sites can be inserted downstream or 3´ of the reporter gene to study how microRNAs affect RNA stability.
How is Reporter Gene Activity Measured?
Reporter genes or rather the proteins translated from the reporter gene sequence can be assayed by detecting enzymatic activity or spectrophotometric characteristics, or indirectly with antibody-based assays. In general, enzymatic assays are more sensitive due to the small amount of reporter enzyme required to generate detectable levels of reaction products. A potential limitation of enzymatic assays is high background if there is endogenous enzymatic activity in the cell (e.g., β-galactosidase breaks down lactose). Antibody-based assays are generally less sensitive but will detect the reporter protein whether it is enzymatically active or not.
Fundamentally, a reporter assay uses a protein to measure a biological outcome using an observable parameter like bioluminescence.
Bioluminescent Reporter Genes
Why Choose Luminescence Over Fluorescence?
Photons are produced primarily through chemiluminescence and fluorescence as a consequence of energy transitions from excited-state molecular orbitals to lower energy orbitals. However, the excited-state orbitals are created differently. In chemiluminescence, the excited states are the product of exothermic chemical reactions, whereas in fluorescence the excited states are created by absorption of light.
This difference in excited state creation greatly affects the assay character. For instance, fluorescence-based assays tend to be much brighter because high-intensity light can be used to generate the excited state. In chemiluminescent assays, the chemical reactions usually yield lower rates of photon emission. The greater brightness of fluorescence would appear to correlate with better assay sensitivity, but commonly this is not the case. Assay sensitivity is determined by a statistical analysis of signal relative to background, where the relative signal consists of a sample measurement minus the background measurement. Fluorescent assays tend to have much higher backgrounds, leading to lower relative signals.
The higher background in fluorescent assays is primarily because fluorometers cannot discriminate perfectly between the very high influx of photons into the sample and the much smaller emission of photons from the analytical fluorophores. This discrimination is accomplished largely by optical filtration—emitted photons have longer wavelengths than excitation photons—and by geometry because emitted photons typically travel a different path than excitation photons. However, optical filters are not perfect in their ability to differentiate between wavelengths, and photons can change directions through scattering. Chemiluminescence has the advantage that, because photons are not required to create the excited states, they do not constitute an inherent background when measuring photon efflux from a sample. The resulting low background permits precise measurement of very small changes in light.
Learn more about the bioluminescence advantage in this article.
Available Bioluminescent Reporter Genes
Bioluminescence is a natural phenomenon of living organisms creating their own light. The basis of bioluminescence is the interaction of the enzyme luciferase with a luminogenic substrate (e.g., luciferin) to produce light (Figure 1). The luciferases that are used most widely are beetle luciferases (including firefly luciferase), Renilla luciferase and a modified deep sea shrimp luciferase (NanoLuc® luciferase). Luciferase genes have been cloned from bacteria, beetles (e.g., firefly), Renilla, deep sea shrimp (Oplophorus), Aequorea, Vargula and Gonyaulax (a dinoflagellate). Of these, only luciferases from bacteria, beetles, deep sea shrimp and Renilla have found general use as reporter genes to assess transcriptional expression. Here we describe commonly used luciferase reporter genes.
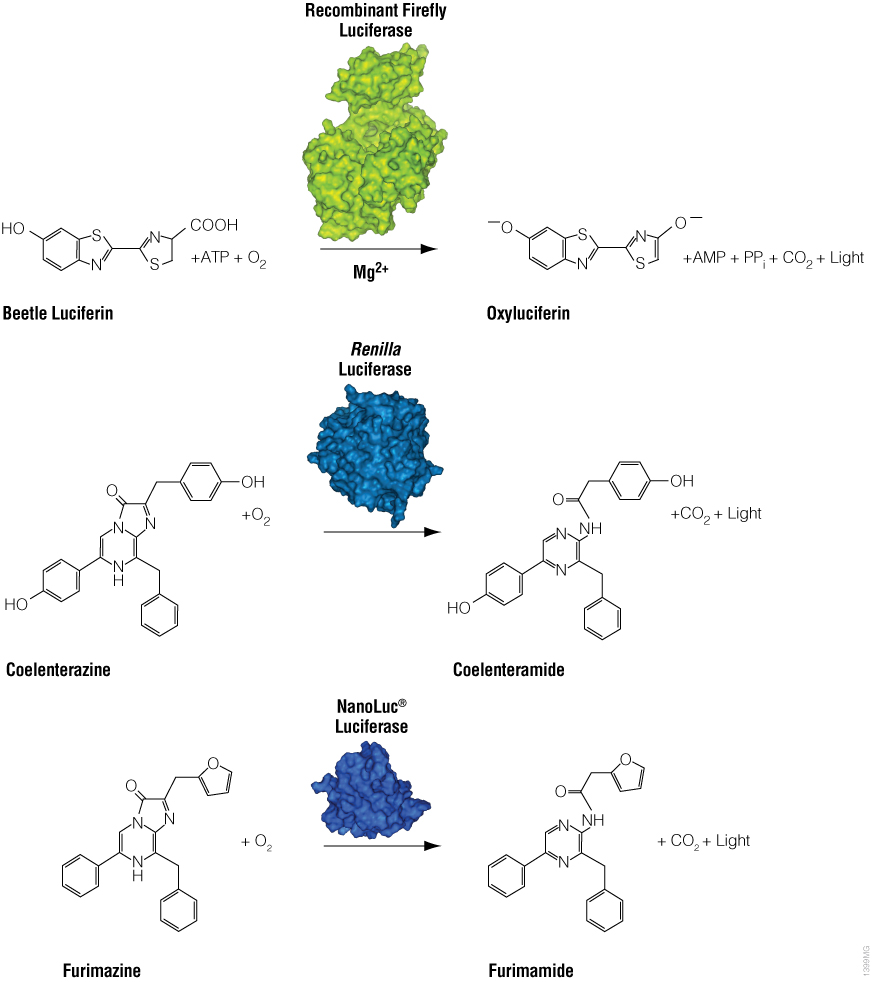
Figure 1. Diagram of firefly, Renilla and NanoLuc® luciferase chemical reactions with their respective substrates (beetle luciferin, coelenterazine and furimazine) to produce light.
Firefly Luciferase
Firefly luciferase is by far the most commonly used bioluminescent reporter. This monomeric enzyme of 61kDa was cloned from the North American firefly (Photinus pyralis) and catalyzes a two-step reaction in which oxidation of D-luciferin yields green to yellow light, typically 550–570nm in the light spectrum (Figure 1). The first reaction step is luciferin activation with ATP to give a luciferyl-adenylate and pyrophosphate. In the second step, the luciferyl-adenylate reacts with molecular oxygen to yield oxyluciferin in an electronically excited state and CO2. The excited-state oxyluciferin then returns to the ground state with the release of light. When mixed with chemical substrates, firefly luciferase produces an initial burst of light that decays over about 15 seconds to a low level of sustained luminescence.
To stabilize luminescence and make the assay more convenient for routine laboratory use, coenzyme A (CoA) is incorporated to yield maximal luminescence intensity that slowly decays over several minutes. An optimized assay containing coenzyme A generates relatively stable luminescence in less than 0.3 seconds, with linearity over a 100-millionfold range of enzyme concentrations and sensitive enough to quantitate fewer than 10–20 moles of enzyme.
The popularity of native firefly luciferase as a genetic reporter is due to the sensitivity and convenience of the enzyme assay and tight coupling of protein synthesis with enzyme activity. Firefly luciferase, which is encoded by the luc gene, is a monomer that does not require any post-translational modifications and becomes a mature enzyme directly upon translation of its mRNA. Catalytic activity is available immediately after release from the ribosome. Also, firefly luciferase has a relatively short half-life in cells compared to other commonly used reporters.
NanoLuc® Luciferase
NanoLuc® luciferase is a small monomeric enzyme (19.1kDa, 171 amino acids) based on the luciferase from the deep sea shrimp Oplophorus gracilirostris (Hall et al. 2012). This engineered enzyme uses a novel substrate, furimazine, to produce high-intensity, glow-type blue-light luminescence (λmax = 465nM) in an ATP-independent reaction (Figure 1). The signal half-life is >2 hours, and luminescence is about 100-fold brighter than that of firefly or Renilla luciferase (Figure 2). See why the brightness of NanoLuc® Luciferase matters in this chalk talk:

In mammalian cells, NanoLuc® luciferase shows no evidence of post-translational modifications, disulfide bonds or subcellular partitioning. The enzyme is active over a broad pH range (pH 6–8), exhibits high physical stability and retains activity at temperatures of up to 55°C or in culture medium for >15 hours at 37°C.
Unlike other forms of luciferase, NanoLuc® luciferase is ideally suited for both standard (lytic) and secretion-based (nonlytic) reporter gene applications. The small size of the gene (513bp) and encoded protein is ideal for reporter virus applications and protein fusions. In addition, live-cell substrates can be used for imaging applications.
In addition, the NanoLuc® luciferase sequence can be fused to another gene to create a fusion protein. These fusions can be expressed in cells for use in many applications, including as a bioluminescent donor in bioluminescence resonance energy transfer (BRET).
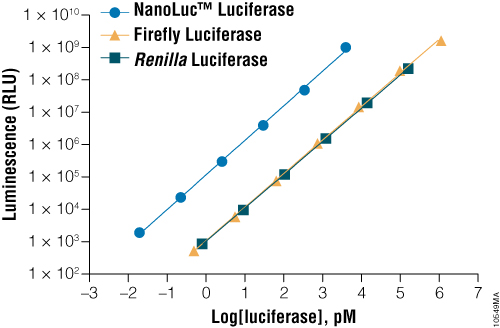
Figure 2. A comparison of the sensitivity of NanoLuc®, firefly and Renilla luciferase assays. Luminescence was measured from varying concentrations of purified luciferases after mixing the reporter enzyme with its respective detection reagent. NanoLuc® luciferase was approximately 100-fold brighter than firefly or Renilla luciferases at equivalent concentrations. Luminescence was measured using the Nano-Glo® Luciferase Assay for NanoLuc® luciferase, ONE-Glo™ Luciferase Assay System for firefly luciferase and Renilla-Glo™ Luciferase Assay System for Renilla luciferase.
Renilla Luciferase
Renilla luciferase is a 36kDa monomeric enzyme that catalyzes the oxidation of coelenterazine to yield coelenteramide and blue light of 480nm (Figure 1). The host organism, Renilla reniformis (sea pansy), is a coelenterate that creates bright green flashes upon tactile stimulation, apparently to ward off potential predators. The green light is created through association of the luciferase with a green fluorescent protein and represents a natural example of BRET.
As a reporter molecule, Renilla luciferase, which is encoded by the Rluc gene, provides many of the same benefits as firefly luciferase. Historically, the presence of nonenzymatic luminescence, termed autoluminescence, resulted in high background and reduced assay sensitivity; however, improvements in assay chemistry have nearly eliminated this problem. In addition, the simplicity of the Renilla luciferase chemistry and, more recently, improvements to the luciferase substrate have meant Renilla luciferase can be quantitated in living cells in situ or in vivo.
Optimizing Reporter Gene Activity
An ideal genetic reporter should: i) express uniformly and optimally in the host cells; ii) only respond to effectors that the assay intends to monitor (avoid anomalous expression); and iii) have a low intrinsic stability to quickly reflect transcriptional dynamics. Despite their biology and enzymology, native luciferases are not necessarily optimal as genetic reporters. That is why Promega scientists have made significant improvements in luciferase expression, reducing the risk of anomalous expression and destabilizing these reporters. The key strategies to achieve these improvements are described here.
Removing the Peroxisomal Targeting Site
Normally, in the firefly light organ, luciferase is located in specialized peroxisomes of photocytic cells. When the firefly enzyme is expressed in foreign hosts, a conserved translocation signal (C-terminal tripeptide sequence, -Ser-Lys-Leu) causes luciferase to accumulate in peroxisomes and glyoxysomes, which may interfere with normal cellular physiology and performance of the genetic reporter.
To develop an optimal cytoplasmic form of the luciferase gene, our scientists replaced the existing C-terminal sequence with a new C-terminal sequence, -Ile-Ala-Val. This modified luciferase has consistently yielded about four- to fivefold greater luminescence than the native enzyme when expressed in NIH/3T3 cells. Renilla and NanoLuc® luciferases do not contain a targeting sequence and are not affected by peroxisomal targeting.
Optimizing Codons
Although redundancy in the genetic code means amino acids are encoded by multiple codons, different organisms favor some codons over others. The efficiency of protein translation in a non-native host cell can be increased substantially by adjusting the codon usage frequency while maintaining the same protein composition. The native luciferase genes cloned from firefly, sea pansy (Renilla reniformis) and deep sea shrimp use codons that are not optimal for expression in mammalian cells. Therefore, Promega scientists systematically altered the codons to the preferred ones while removing inappropriate or unintended transcription regulatory sequences used in mammalian cells. As a result, luciferase expression levels significantly increased, up to several hundredfold in some cases (Figures 3 and 4).
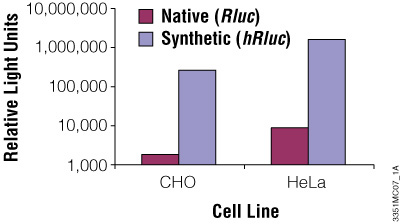
Figure 3. The synthetic Renilla luciferase gene supports higher expression than the native gene in mammalian cells. CHO and HeLa cells were transfected with the pGL3-Control Vector (Cat.# E1741; contains the SV40 enhancer/promoter) harboring either the synthetic hRluc or native Rluc gene. Cells were harvested 24 hours after transfection and Renilla luciferase activity assayed using the Dual-Luciferase® Reporter Assay System.
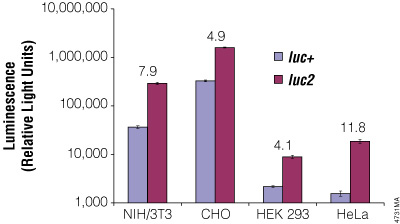
Figure 4. The firefly luc2 gene displays higher expression than the luc+ gene. The luc2 gene was cloned into the pGL3-Control Vector, replacing the luc+ gene. Thus both firefly luciferase genes were in the same pGL3-Control Vector backbone. The two vectors containing the firefly luciferase genes were co-transfected into NIH/3T3, CHO, HEK 293 and HeLa cells using the phRL-TK Vector as a transfection control. Twenty-four hours post-transfection the cells were lysed with Passive Lysis 5X Buffer (Cat.# E1941), and luminescence was measured using the Dual-Luciferase® Reporter Assay System (Cat.# E1910). Firefly luciferase luminescence (in relative light units) was normalized to Renilla luciferase expression from the phRL-TK Vector. The increase in expression of luc2 compared to luc+, indicated as the fold increase, for each cell line is listed above each pair of bars.
Removing Cryptic Regulatory Sequences
The presence of cryptic regulatory sequences in a reporter gene may adversely affect transcription, resulting in anomalous expression of the reporter. A cryptic regulatory sequence can be a transcription factor-binding site or a promoter module (defined as at least two transcription factor-binding sites separated by a spacer; Klingenhoff et al. 1999). Additionally, an enhancer element can elevate levels of transcription in the absence of a promoter sequence or increase basal levels of gene expression in the presence of transcription regulatory sequences. These regulatory sequences can increase or decrease transcription, affecting overall gene expression (Klingenhoff et al. 1999).
The cryptic regulatory sequences in the luc gene were removed without changing the amino acid sequence to create the luc2 gene. In addition, sequences resembling splice sites, poly(A) addition sequences, Kozak sequences (translation start sites for mammalian cells), E. coli promoters or E. coli ribosome-binding sites were removed wherever possible. This process has led to a greatly reduced number of cryptic regulatory sequences (Figure 5) and therefore a reduced risk of anomalous transcription. A similar process was performed using Renilla luciferase to produce the hRluc gene.
Figure 5. Reduced number of consensus transcription factor-binding sites for the luc2 gene. The number of consensus transcription factor-binding sites identified in the luc+ gene is greatly reduced in the luc2 gene.
Adding Degradation Signals
When performing reporter assays, the total accumulated reporter protein within cells is measured. This accumulation occurs over the intracellular lifetime of the reporter, which is determined by both protein and mRNA stability. A dynamic reporter will better reflect transcriptional changes of both upregulated and downregulated gene expression.
NanoLuc® luciferase is more stable than firefly and Renilla luciferase reporter proteins. Due to protein stability, reporter response may lag behind changes in the underlying transcriptional events by several hours. To reduce the cellular half-life of the reporter gene and improve reporter responsiveness, Promega scientists developed destabilized luciferase reporters by genetically fusing a protein degradation sequence to the luciferase reporter gene (Li et al. 1998). After evaluating many degradation sequences for their effect on response rate and signal magnitude, Promega scientists chose two sequences to reduce the reporter half-life: the PEST protein degradation sequence and a sequence composed of two protein degradation sequences (CL1 and PEST). The luc2 gene with a PEST sequence (luc2P) and CL1 and PEST sequences (luc2CP), named the Rapid Response™ genes, respond more rapidly to stimuli but at the expense of signal intensity. The luc2P gene (approximately 1-hour protein half-life; half-life varies by cell line) responds much more rapidly than luc2 (approximately 3-hour half-life), with moderate signal intensity. The luc2CP gene (approximately 0.4-hour half-life) responds more quickly and has lowest signal intensity. The NanoLuc® gene product with a PEST sequence (NlucP) has a protein half-life of about 20 minutes but still maintains a bright signal due to the enzyme's higher specific activity. These destabilized reporters respond faster and often display greater signal-to-background compared to nondestabilized counterparts.
Adding a Secretion Signal
Native Oplophorus luciferase catalyzes the luminescent oxidation of the substrate coelenterazine to produce blue light. The enzyme is secreted by the shrimp as a defense mechanism against predation. In the laboratory, a secreted reporter is useful for extracellular detection—for example, to avoid cell lysis and maintain cell viability. To maximize the protein secretion efficiency in mammalian cells, Promega scientists replaced the native secretion signal with a mammalian protein secretion signal, resulting in the secNluc gene. Secreted NanoLuc® luciferase is ideal for nondestructive reporter gene assays in cultured cells due to the enzyme's thermostability, meaning the enzyme can accumulate in the extracellular medium.
Reporter Gene Vectors
Vectors are plasmids, extrachromosomal elements, that carry reporter gene sequences for delivery into cultured cells. Not only do vectors provide the reporter gene, but may also have multiple cloning sites, promoters, response elements, polyadenylation sequences, mammalian selectable markers or other sequence elements that help with expression and selection. Reporter vectors are critical for success in reporter assays.
pGL4 Luciferase Reporter Vectors Encoding Firefly and Renilla Luciferase
Because vectors are used to deliver the reporter gene to host cells, regulatory sequences such as transcription factor-binding sites and promoter modules within the vector backbone can lead to high background and anomalous responses. This is a common issue for mammalian reporter vectors, including the pGL3 Luciferase Reporter Vectors. Our scientists applied the successful "cleaning" strategy described for reporter genes to the entire pGL3 Vector backbone, removing cryptic regulatory sequences wherever possible, while maintaining reporter functionality. In addition, the multiple cloning region was redesigned with a unique SfiI site for easy transfer of the DNA element of interest, the f1 origin of replication removed and an intron deleted. Furthermore, a synthetic poly(A) signal/transcriptional pause site was placed upstream of either the multiple cloning region (in promoterless vectors) or HSV-TK, CMV or SV40 promoter (in promoter-containing vectors). This extensive effort resulted in the totally redesigned and unique vector backbone of the pGL4 Vectors. There are also a series of pGL4 Vectors with partial deletions of the CMV promoter for nuanced usage of the strong promoter.
The pGL4 family of luciferase vectors incorporates a variety of features such as your choice of optimized firefly or Renilla luciferase genes, Rapid Response™ versions for improved temporal response, mammalian selectable markers, basic vectors without promoters, promoter-containing control vectors and predesigned vectors with your choice of several response elements (Figure 6).

Figure 6. The family of pGL4 Luciferase Reporter Vectors incorporates a variety of additional features, such as a choice of luciferase genes, Rapid Response™ versions, a variety of mammalian selectable markers and vectors with or without promoters and response elements.
pNL Vectors Encoding NanoLuc® Luciferase
NanoLuc® luciferase is available in a variety of vectors for use in reporter gene assays of transcriptional control. These pNL vectors are based on the pGL4 vector backbone and thus offer many of the same advantages: Removal of transcription factor-binding sites and other potential regulatory elements to reduce the risk of anomalous results, easy sequence transfer from existing plasmids and a choice of several promoter sequences, including no promoter, minimal promoter and viral promoters. The family of vectors offer a choice of NanoLuc® genes (unfused Nluc, PEST-destabilized NlucP and secreted secNluc). These NanoLuc® gene variations are codon-optimized and have had many potential regulatory elements or other undesirable features such as common restriction enzyme sites removed.
In addition, there are coincidence reporter vectors that encode both NanoLuc® and firefly luciferases on a single transcript. These vectors come with no promoter, a minimal promoter or CMV promoter for use in high-throughput screening. Study the rate of protein turnover of two key signaling proteins involved in response to cellular stress using reporter vectors.
Reporter Vectors
Our extensive line of state-of-the-art bioluminescent reporter vectors includes the pGL4 Vectors —the latest generation of firefly and Renilla luciferase reporter vectors, and the pNL Vectors, which contain the NanoLuc® luciferase reporter gene.
Measuring Reporter Gene Activity
The basis of bioluminescent detection assays is harnessing the efficient light-emitting chemistry of the enzyme and its substrate for use in analytical methods. By holding the concentration of each reaction component constant, except for one component that varies in relation to the process being studied, the resulting light is directly proportional to the variable component. This couples an observable parameter to the reaction outcome. In assays using luciferase, the variable component may be the luciferase itself, its substrates or cofactors. Due to the very low backgrounds in bioluminescence, the proportional linear range can be enormous, typically extending 104- to 108-fold over the concentration of the variable component.
Intracellular luciferase is typically quantified by adding a buffered solution containing detergent to lyse the cells and a luciferase substrate to initiate the luminescent reaction. The luminescence slowly decays, decreasing the signal over time. To maintain steady luminescence of firefly and Renilla luciferase assays over an extended period of time, ranging from minutes to hours, luminescent reaction needs to be inhibited to various degrees. Even under these conditions, as few as 10–20 moles of luciferase or less per sample may be quantified. This corresponds to roughly 10 molecules per cell.
In contrast, NanoLuc® luciferase has an inherently longer signal half-life. NanoLuc®-based assays, which use a novel furimazine substrate to measure NanoLuc® luciferase activity, exhibit a much brighter signal and a much slower rate of luminescence decay, with a signal half-life of approximately 120 minutes. These extended half-life assays are convenient for reporter gene applications. Simply add the reagent, and measure the resulting luminescence.
Most reporter gene assays involve either a one reporter gene or two. Most often, the second reporter is expressed from a "control" vector to normalize results of the experimental reporter. For example, the second reporter can control for variation in cell number or transfection efficiency. Typically, the control reporter gene is driven by a constitutive promoter, and the control vector is cotransfected with the "experimental" vector. Different reporter genes are used for the control and experimental vectors so that the relative activities of the two reporter products can be assayed individually. The Introduction to Reporter Gene Assays animation below demonstrates the basic design of a reporter assay using the Dual-Luciferase® Reporter Assay System as an example to study promoter structure, gene regulation and signaling pathways.

Alternatively, you can design dual-reporter assays in which both reporter genes are used as experimental reporters. Such dual-reporter assays can be particularly useful for efficiently extracting more information in a single experiment. For more information on using one or two experimental reporters in a dual-luciferase assay, read the article on how to choose a NanoDLR™ Dual Reporter Assay format.
The following section provides information about specific bioluminescent reporter assays with information to help you choose the reporter gene and assay that suit your research needs. Tables 1, 2 and 3 summarize available luciferase genes, assays and reagents.
Single-Reporter Assays
Assays based on a single reporter provide the quickest means to acquire gene expression data from cells. However, because cells are inherently complex, the information gleaned from a single-reporter assay may be insufficient to achieve detailed and accurate results. Thus, one of the first considerations when choosing a reporter methodology is deciding if the information from a single reporter is sufficient or if you would benefit from the additional information that can be gleaned from a second reporter (e.g., normalization of differences in transfection efficiency or cell number). If more information is required, see the section below that covers Dual-Reporter Assays.
When choosing a luciferase assay, a trade-off between luminescence intensity and duration is often necessary because bright reactions fade relatively quickly. Using a firefly or Renilla luciferase assay that yields maximum luminescence results in higher sensitivity, but using an assay with a longer signal half-life and a more stable luminescent signal is more convenient when performing assays in multiwell plates. Of the extended half-life firefly luciferase assays, there are the ONE-Glo™ Luciferase Assay System (Cat.# E6110) and ONE-Glo™ EX Luciferase Assay System (Cat.# E8110). The ONE-Glo™ Luciferase Assay System yields maximal luminescence intensity, has a 45-minute half-life and is more tolerant of nonoptimal reaction conditions. The ONE-Glo™ EX Reagent has a longer signal half-life (approximately 2 hours), is more stable at 4°C or room temperature once reconstituted, exhibits less interference from phenol red in cell culture medium and has less odor than other luciferase assay reagents.
Figure 7. Example data that illustrates the luminescent signal stability of various firefly luciferase reporter assays. In this experiment, 100µl purified firefly luciferase (13.8ng/ml) was combined in a 96-well plate with 100µl of Bright-Glo™, ONE-Glo™, ONE-Glo™ EX, Dual-Glo® Luciferase or Steady-Glo® Reagents. Luminescence was measured periodically over 2 hours, n = 8.
The Steady-Glo® Luciferase Assay System provides even longer luminescence duration but with lower intensity. The Steady-Glo® Reagent is added directly to the culture medium for mammalian cells, so prior cell lysis is not necessary. This allows you to grow cells in multiwell plates, and then measure reporter expression in a single step. Renilla luciferase assays with different signal intensities and half-lives are also available.
Sacrificing luminescence intensity for signal half-life is less of a concern with NanoLuc® luciferase. The Nano-Glo® Luciferase Assay System (Cat.# N1110) provides a simple, single-addition reagent that generates a glow-type signal with a half-life of approximately 120 minutes in commonly used tissue culture media. The reagent contains an integral lysis buffer for direct use with cells expressing NanoLuc® luciferase, but is also compatible with culture medium containing secreted luciferase.
Dual-Reporter Assays
Measuring two reporters in a single assay is called a dual-reporter assay or, if both reporters are luciferases, a dual-luciferase assay. While the most commonly used dual-reporter assays measure both firefly and Renilla luciferase activities, the next-generation dual-luciferase assay uses NanoLuc and firefly luciferases. These pairs of luciferases use different substrates and thus can be differentiated by their enzymatic specificities. Performing most dual-luciferase assays involves adding two reagents to each sample and measuring luminescence following each addition. Adding the first reagent activates the first luciferase reporter reaction; adding the second reagent extinguishes first luciferase reporter activity and initiates the second luciferase reaction.
The Nano-Glo® Dual-Luciferase® Reporter Assay System (Cat.# N1610) measures firefly and NanoLuc® luciferase activity. This combination of reporters and assay has several advantages, including lower background levels due to better quenching of firefly luciferase activity and higher sensitivity due to brighter NanoLuc® luminescence. The long signal half-lives (approximately 2 hours for firefly luciferase and NanoLuc® luciferase) make this assay particularly useful for high-throughput screening. In addition, the lower firefly luciferase background and brighter NanoLuc® signal allows greater flexibility in assay configuration. Either NanoLuc® or firefly luciferase can be used as the experimental reporter, with the other luciferase used as the normalization control, or both reporter genes can be used as the experimental reporter, providing you with more information from a single set of assays.
The older technology dual-reporter assay is the Dual-Luciferase® Reporter Assay System (Cat.# E1910), which measures both firefly and Renilla luciferase activities sequentially from a single sample. This system requires cell lysis prior to performing the assay and requires the use of reagent injectors with multiwell plates.
The Dual-Glo® Luciferase Assay System also measures both firefly and Renilla luciferase activities from a single sample and provides a longer luciferase signal half-life, which is useful when performing reporter assays in multiwell plates. As with other reagents designed for use in multiwell plates, the Dual-Glo® Assay works directly in mammalian cell culture medium without prior cell lysis.
In general, dual-reporter assays improve experimental accuracy and efficiency by: i) reducing variability that can obscure meaningful correlations; ii) normalizing interfering phenomena that may be inherent in the experimental system; and iii) normalizing differences in transfection efficiencies between samples. In addition, dual-reporter assays can reduce the number of nonrelevant hits (i.e., "false" hits) due to direct interaction of compounds with the reporter protein in high-throughput screenings. The use of co-incidence reporters—reporters that have dissimilar profiles of compound interference—can help differentiate compounds that modulate the biological pathway of interest from those that affect the stability or activity of the reporter enzyme.
Reducing Variability
Because cells are complex micro-environments, significant variability can occur between samples within an experiment and between experiments performed at different times. Challenges include maintaining uniform cell density and viability between samples and accomplishing reproducible transfection of exogenous DNA. The use of multiwell plates introduce variables such as edge effects, which are brought about by differences in heat distribution and humidity across a plate. Dual-reporter assays can control for much of this variability, leading to more accurate and meaningful comparisons between samples (Hawkins et al. 2002; Hannah et al. 1998; Wood, 1998; Faridi et al. 2003).
Live-Cell Substrates
Researchers strive to monitor cellular activities with as little effect on the cell as possible. Most reporter activity assays use an endpoint lytic method to disrupt cells so that the environment surrounding the reporter enzyme can be carefully controlled. However, there are advantages to using a nonlytic assay to measure reporter gene activity, including continuous monitoring of expression changes over time and multiplexing with assays that assess cell health. Promega scientists have developed a variety of live-cell substrates to monitor luciferase activity without disrupting cells.
With a single-reagent addition to culture medium, NanoLuc® luciferase activity can be monitored for hours or even days. The Nano-Glo® Live Cell Assay System can monitor reporter activity for up to 2 hours with the greatest light intensity. For longer times, the Nano-Glo® Vivazine® Live Cell Substrate can monitor NanoLuc® activity over several hours, but with a decreased light intensity while the Nano-Glo® Endurazine™ Live Cell Substrate has the greatest signal stability for monitoring reporter activity for days, but with the lowest signal intensity of the NanoLuc® substrates. These live-cell detection reagents can provide kinetic measurements of reporter expression for investigating protein interaction and simplifying time course studies.
Renilla luciferase requires only oxygen and coelenterazine to generate luminescence, providing a simple luciferase system to measure luminescence from living cells. The EnduRen™ and ViviRen™ Live Cell Substrates overcome the instability of coelenterazine in aqueous solution and easily generate luminescence from live cells expressing Renilla luciferase. Because the cells are still alive, you can determine viable cell number by multiplexing with another assay.
An alternative to live-cell substrates is a secreted form of reporter protein, which can be quantified by measuring reporter activity in the cell culture medium. NanoLuc® luciferase fused to an N-terminal secretion signal provides a secreted form (secNluc) that does not require cell lysis prior to or during the reporter assay.
Reporter Assay Resources
- Single-Reporter Assays:
Detection of HIF1α Expression at Physiological Levels Using NanoLuc® Luciferase and the GloMax® Discover System - Dual-Reporter Assays:
Choosing a Luciferase Reporter Assay: A Comparison Guide
Designing a Bioluminescent Reporter Assay: Choosing Your Experimental Reporter
Designing a Bioluminescent Reporter Assay: Normalization
Mix and Match Primary and Control Reporters using the NanoDLR™ Dual-Luciferase Assay System
Reporter Assay Products
Promega reporter assays provide a wide range of choices for single or dual-reporter formats.
Reporter Gene Applications
The conventional use of reporter genes is largely to analyze gene expression and dissect the function of cis-acting genetic elements such as promoters and enhancers (so-called "promoter bashing"). In typical experiments, deletions or mutations are made in a promoter region, and their effects on coupled expression of a reporter gene are quantitated. However, reporter genes also can be used to study other cellular events, including events that are not related to gene expression such as cell health and signaling pathways. For more information, view the following Introduction to Bioluminescent Assays animation.

Normalize for Changes in Cell Physiology
Events associated with cell physiology can affect reporter gene expression. Of particular concern is the effect of cytotoxicity, which can mimic genetic downregulation when using a single-reporter assay. Reporter assays that can be multiplexed with a cell viability or cytotoxicity assay for independent monitoring of both reporter expression and cell viability to avoid data misinterpretation (Farfan et al. 2004). The use of multiplexed assays allows correlation of events within cells, such as the coupling of target suppression by RNAi, to the consequences on cellular physiology (Hirose et al. 2002).
Luminescent cell viability or cytotoxicity assays, such as the CellTiter-Glo® 2.0 Cell Viability Assay (Cat.# G9241) and CytoTox-Glo™ Cytotoxicity Assay (Cat.# G9290), use a stabilized firefly luciferase to generate a luminescent signal that indicate cell health. Because these assays contain firefly luciferase, they cannot be directly combined with a firefly luciferase reporter assay. However, the assays can be readily combined with nondestructive luciferase assays. For example, quantitating NanoLuc® luciferase expression by adding one of the Nano-Glo® Live Cell Substrates to the culture medium and measuring luminescence. When the reporter measurements are completed, the CellTiter-Glo® 2.0 or CytoTox-Glo™ Reagent is added to the sample to inactivate NanoLuc® luciferase and initiate luminescence to assess cell viability. Or measure the activity of the secreted NanoLuc® reporter from an aliquot of culture medium using the Nano-Glo® Luciferase Assay System.
Alternatively, you can multiplex a luminescent reporter assay with fluorescent cell viability and cytotoxicity assays to monitor cell health and normalize single-reporter assay results. For example, the CellTiter-Fluor™ Cell Viability Assay (Cat.# G6080) is a nonlytic, fluorescence assay that measures the relative number of viable cells in a population. The CellTox™ Green Cytotoxicity Assay (Cat.# G8741) uses a proprietary dye that is excluded from viable cells but preferentially binds to DNA from dead cells. Upon DNA binding, fluorescence of the dye is substantially enhanced, and the resulting fluorescence is proportional to the level of cytotoxicity. These cell health assays are well suited for multiplexing with homogeneous luciferase assay reagents such as Bright-Glo™, Steady-Glo® and ONE-Glo™ Luciferase Assay Systems (Zakowicz et al. 2008), the Renilla-Glo® Luciferase Assay System as well as the Nano-Glo® Live Cell Substrates.
Additional Resources for Reporter Assays and Cell Health
Monitor RNA Interference
RNA interference (RNAi) is a phenomenon by which double-stranded RNA complementary to a target mRNA can specifically inactivate a gene by stimulating degradation of the target mRNA. As such, RNAi or more generally RNA silencing, has emerged as a powerful tool to analyze gene function.
Bioluminescent reporters have been harnessed to study RNAi. For example, the pmirGLO Dual-Luciferase miRNA Target Expression Vector (Cat.# E1330) is based on dual-luciferase technology, with firefly luciferase as the primary reporter to monitor mRNA regulation and Renilla luciferase as a control reporter for normalization. The pmirGLO Vector quantitatively evaluates microRNA (miRNA) activity by inserting miRNA target sites downstream or 3´ of the firefly luciferase gene. Reduced firefly luciferase expression indicates binding of endogenous or introduced miRNAs to the cloned miRNA target sequence.
Assess Cell Signaling Pathways
Luciferase reporter assays are widely used to investigate cellular signaling pathways and as high-throughput screening tools for drug discovery (Brasier et al. 1992, Zhuang et al. 2006). Synthetic constructs with cloned regulatory elements directing reporter gene expression can be used to monitor signal transduction and identify the signaling pathways involved. By linking luciferase expression to specific response elements (REs) within the reporter construct, transfecting cells with this construct, adding a particular treatment, and then measuring reporter activity, researchers can determine what REs are used, and thus, what signaling pathways are involved. The use of inhibitors and siRNAs can be used to confirm what factors are involved in this response.
There are a variety of firefly luciferase pGL4 Vectors with your choice of a number of response elements and regulatory sequences for use in characterizing and modulating signaling pathways. For example, CREB, CRE and STAT5RE are available for your research. See Table 1 for a complete list. Many of these vectors encode the hygromycin-resistance gene to allow selection of stably transfected cell lines.
Examine Nuclear Receptors
Bioluminescent reporter genes can also characterize nuclear receptors, a class of ligand-regulated transcription factors that sense the presence of steroids and other molecules inside the cell. Nuclear receptors typically reside in the cytoplasm and are often complexed with associated regulatory proteins. Ligand binding triggers translocation into the nucleus, where the receptors bind specific response elements via the DNA-binding domain, leading to upregulation of the adjacent gene. Bioluminescent reporters can be harnessed to identify and characterize nuclear receptor agonists, antagonists, co-repressors and co-activators using a universal receptor assay. The universal nuclear reporter assay can be thought of as a "one-hybrid" assay, where the ligand-binding domain (LBD) of a nuclear receptor is fused to yeast GAL4 transcription factor and when a ligand binds to the nuclear receptor, firefly luciferase is expressed (Figure 8).

Figure 8. The universal nuclear receptor assay. The ligand-binding domain of the nuclear receptor is fused to GAL4. Within the cell, binding of the appropriate ligand to the nuclear receptor-GAL4 fusion protein releases any co-repressors bound to the ligand-binding domain. Co-activators help recruit the transcription machinery to the luciferase reporter gene, resulting in luciferase expression and an increase in luminescence.
To use the universal nuclear receptor assay, simply cotransfect the cell line of interest with a construct encoding the LBD-GAL4 DBD fusion protein and a suitable reporter vector like pGL4.35[luc2P/9XGAL4UAS/Hygro] Vector (Cat.# E1370) that has multiple copies of the GAL4 upstream activation sequence (UAS) upstream of a minimal promoter to drive expression of the firefly luciferase reporter gene. Two to three days post-transfection, treat cells with the test compounds of interest, then measure luciferase activity. This approach allows you to convert any cell line into a nuclear receptor-responsive cell line for identifying receptor agonists, antagonists, co-activators and co-repressors. You can even perform mutagenesis on the ligand-binding domain to determine the effect in your responsive cell line without interference from the endogenous receptor.
To simplify universal nuclear receptor assays, there are additional reagents to use. The pBIND-ERα Vector (Cat.# E1390) contains the yeast Gal4 DBD and an estrogen receptor ligand-binding domain (ER-LBD) gene fusion, and the pBIND-GR Vector (Cat.# E1581) contains the yeast Gal4 DBD and glucocorticoid receptor ligand-binding domain (GR-LBD) gene fusion. Promega also offers the pFN26A (BIND) hRluc-neo Flexi® Vector (Cat.# E1380) for expressing a fusion protein comprised of the GAL4 DBD, a linker segment and an in-frame protein-coding sequence under the control of the human cytomegalovirus (CMV) immediate early promoter. Each BIND vector contains a Renilla luciferase/neomycin resistance co-reporter for normalization of transfection efficiency or construction of a double-stable cell line without the need for additional cloning.
Research GPCRs
Studying G protein-coupled receptors (GPCRs), which regulate a wide-range of biological functions and are one of the most important target classes for drug discovery (Klabunde et al. 2002), can be studied with reporter genes. The firefly luciferase-based GloSensor™ cAMP Assay provides a sensitive and easy-to-use format to interrogate overexpressed or endogenous GPCRs that signal via changes in intracellular cAMP concentration. The assay uses genetically encoded biosensor variants comprised of cAMP-binding domains fused to mutant forms of Photinus pyralis luciferase. cAMP binding induces conformational changes that promote large increases in light output. Following pre-equilibration with the GloSensor cAMP Reagent, cells transiently or stably expressing the biosensor variant can be used to assay GPCR function and kinetically measure cAMP accumulation or turnover in living cells. Moreover, the assay offers a broad dynamic range, with up to 500-fold changes in light output. The sensitive assay detects Gi-coupled receptor activation or inverse agonist activity in the absence of artificial stimulation by compounds such as forskolin. For more information, visit the GloSensor™ Technology page.
Additional Resources for GPCR Reporter Assays
Use Reporter Cell Lines to Study Response Pathways
The GloResponse™ Cell Lines contain optimized luciferase reporter technology integrated into a cell line. These cells use the destabilized and optimized luc2P gene for greater sensitivity and shorter induction times compared to native reporter enzymes. The GloResponse™ NFAT-RE-luc2P HEK293 Cell Line, NFκB-RE-luc2P HEK293 Cell Line and CRE-luc2P HEK293 Cell Line can rapidly and conveniently analyze cell signaling through the NFAT, NF-κB or cAMP response pathways, respectively, by activating a reporter gene. Non-native activators of these pathways, including GPCRs, can be studied after the appropriate proteins are introduced by transfection. These cell lines, generated by clonal selection, provide very high reporter induction levels when the pathway of interest is activated.
GPCR signaling pathways can be categorized into three classes based on the G protein α-subunit involved: Gs, Gi/o and Gq. The GloResponse™ CRE-luc2P HEK293 Cell Line can be used to study and configure screening assays for Gs- and Gi/o-coupled GPCRs, which signal through cAMP and the cAMP response element (CRE). For Gq-coupled GPCRs, which signal through calcium ions and NFAT-RE, use the GloResponse™ NFAT-RE-luc2P HEK293 Cell Line. GPCR assays that use the GloResponse™ Cell Lines are amenable to high-throughput screening. These assays typically have greater response dynamics (fold of induction) than other assay formats and generate high-quality data as indicated by the high Z′ values.
Study Protein Dynamics
The rate of protein turnover is tightly regulated for many signaling proteins. Protein stabilization and subsequent accumulation can occur in response to cell signaling events and changing cellular conditions and result in activation of downstream transcriptional events. The NanoLuc® Stability Sensor Vectors (Cat.# N1381 and Cat.# N1391) enable stability studies of two key signaling proteins, HIF1A and NRF2, and provide a method to directly measure the cellular effects of hypoxia and oxidative stress, respectively (Robers et al. 2014). The vectors encode NanoLuc® luciferase fused to the C terminus of each protein under the control of the CMV promoter. With a constitutive promoter like CMV, changes in light output correlate to dynamic changes in protein levels.
Additional Resources for Studying Protein Dynamics
- Article: Detection of HIF1α expression at physiological levels using NanoLuc® Luciferase and the GloMax® Discover System
- Article: Measuring Intracellular Protein Lifetime Dynamics using NanoLuc® Luciferase
Investigate Protein Interactions
One method used to monitor protein:protein interactions is bioluminescence resonance energy transfer (BRET), where two fusion proteins are made: one fused to the bioluminescent NanoLuc® or Renilla luciferase and another protein fused to a fluorescent molecule. When the two fusion proteins interact, there is an energy transfer from the bioluminescent molecule to the fluorescent molecule, with a concomitant change from blue light to green light (Angers et al. 2000). Using NanoLuc® luciferase as the energy donor and HaloTag® protein labeled with the NanoBRET® 618 fluorophore as the energy acceptor results in an improved BRET assay, the NanoBRET® PPI Assay. The bright, blue-shifted signal from the NanoLuc® donor combined with the far-red-shifted HaloTag® acceptor create a protein interaction assay with optimal spectral overlap, increased signal, and lower background compared to conventional BRET assays.
Another way to detect protein interactions is to use a structural complementation reporter system. That is, when two subunits that are each fused to target proteins of interest and expressed in cells. When the two proteins interact, the subunits come together to form an active enzyme and generate a bright luminescent signal in the presence of substrate. The NanoLuc® Binary Technology (NanoBiT) is composed of Large BiT (LgBiT) and Small BiT (SmBiT) that form the NanoBiT® enzyme when brought together provides a method to follow protein:protein interaction dynamics in real time.
Additional Resources for Investigating Protein Interactions
- Protein:Protein Interactions
- Article: Studying Protein Interactions with BRET and NanoLuc® Luciferase
- Poster: Monitoring intracellular protein interactions using NanoLuc® Binary Technology (NanoBiT)
Image Cells and Whole Animals
Luciferase reporter genes can be used as light-emitting reporters in cellular and animal models. Visualize reporter expression using live-cell luciferase substrates or secreted forms of luciferase for nondestructive, quantitative assays and multiple measures of the same samples without perturbation. NanoLuc® luciferase is well-suited for use in bioluminescence imaging. Its small size means NanoLuc® is less likely to perturb the normal functionality of the fusion partner and bright luminescence requires a few seconds of exposure time to monitor subcellular localization. Learn more about imaging.
Related Products and Resources
Categories
References
- Angers, S. et al. (2000) Detection of β2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proc. Natl. Acad. Sci. USA 97, 3684–9.
- Brasier, A.R. and Ron, D. (1992) Luciferase reporter gene assay in mammalian cells. Methods Enzymol. 216, 386–97.
- Farfan, A. et al. (2004) Multiplexing homogeneous cell-based assays Cell Notes 10, 15–8.
- Faridi, J. et al. (2003) Expression of constitutively active Akt-3 in MCF-7 breast cancer cells reverses the estrogen and tamoxifen responsivity of these cells in vivo. Clin. Can. Res. 9, 2933–9.
- Hall, M.P. et al. (2012) Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 7, 1848–57.
- Hannah, R. et al. (1998) Rapid luciferase reporter assay systems for high-throughput studies. Promega Notes 65, 9–14.
- Hawkins, E.H. et al. (2002) Dual-Glo™ Luciferase Assay System: Convenient dual-reporter measurements in 96- and 384-well plates. Promega Notes 81, 22–6.
- Hirose, F. et al. (2002) Drosophila Mi-2 negatively regulates dDREF by inhibiting its DNA-binding activity. Mol. Cell. Biol. 22, 5182–93.
- Klabunde, T. and Hessler G. (2002) Drug design strategies for targeting G-protein-coupled receptors. Chembiochem. 4, 928–44.
- Klingenhoff, A. et al. (1999) Functional promoter modules can be detected by formal models independent of overall nucleotide sequence similarity. Bioinformatics 15, 180–6.
- Li, X. et al. (1998) Generation of destabilized green fluorescent protein as a transcription reporter. J. Biol. Chem. 273, 34970–5.
- Robers, M. et al. (2014) Measuring intracellular protein lifetime dynamics using NanoLuc® luciferase. PubHub.
- Wood, K.V. (1998) The chemistry of bioluminescent reporter assays. Promega Notes 65, 14–20.
- Zakowicz, H. et al. (2008) Measuring cell health and viability sequentially by same-well multiplexing using the GloMax ® -Multi Detection System. Promega Notes 99, 25–8.
- Zhuang, F. and Liu, Y.H. (2006) Usefulness of the luciferase reporter system to test the efficacy of siRNA. Methods Mol. Biol. 342, 181–7.
Appendix: Vector and Reporter Assay Listings
| Table 1. pGL4 Luciferase Reporter Vectors. | |||||
| Vector | Reporter Gene | Multiple Cloning Region | Protein Degradation Sequence | Gene Promoter | Mammalian Selectable Marker |
|---|---|---|---|---|---|
| pGL4.10 | luc2 | Yes | No | No | No |
| pGL4.11 | luc2P | Yes | hPEST | No | No |
| pGL4.12 | luc2CP | Yes | CL1-hPEST | No | No |
| pGL4.13 | luc2 | No | No | SV40 | No |
| pGL4.14 | luc2 | Yes | No | No | Hygro |
| pGL4.15 | luc2P | Yes | hPEST | No | Hygro |
| pGL4.16 | luc2CP | Yes | CL1-hPEST | No | Hygro |
| pGL4.17 | luc2 | Yes | No | No | Neo |
| pGL4.18 | luc2P | Yes | hPEST | No | Neo |
| pGL4.19 | luc2CP | Yes | CL1-hPEST | No | Neo |
| pGL4.20 | luc2 | Yes | No | No | Puro |
| pGL4.21 | luc2P | Yes | hPEST | No | Puro |
| pGL4.22 | luc2CP | Yes | CL1-hPEST | No | Puro |
| pGL4.23 | luc2 | Yes | No | minP | No |
| pGL4.24 | luc2P | Yes | hPEST | minP | No |
| pGL4.25 | luc2CP | Yes | CL1-hPEST | minP | No |
| pGL4.26 | luc2 | Yes | No | minP | Hygro |
| pGL4.27 | luc2P | Yes | hPEST | minP | Hygro |
| pGL4.28 | luc2CP | Yes | CL1-hPEST | minP | Hygro |
| pGL4.29 | luc2P | No | hPEST | minP + CRE | Hygro |
| pGL4.30 | luc2P | No | hPEST | minP + NFAT RE | Hygro |
| pGL4.31 | luc2P | No | hPEST | adenovirus major late + GAL4 UAS | Hygro |
| pGL4.32 | luc2P | No | hPEST | minP + NF-kB RE | Hygro |
| pGL4.33 | luc2P | No | hPEST | serum response element | Hygro |
| pGL4.34 | luc2P | No | hPEST | SRF RE | Hygro |
| pGL4.35 | luc2P | No | hPEST | GAL4 UAS | Hygro |
| pGL4.36 | luc2P | No | hPEST | murine mammary tumor virus long terminal repeat | Hygro |
| pGL4.37 | luc2P | No | hPEST | minP + antioxidant RE | Hygro |
| pGL4.38 | luc2P | No | hPEST | minP + p53 RE | Hygro |
| pGL4.39 | luc2P | No | hPEST | minP + ATF6 RE | Hygro |
| pGL4.40 | luc2P | No | hPEST | minP + metal RE | Hygro |
| pGL4.41 | luc2P | No | hPEST | minP + heat shock RE | Hygro |
| pGL4.42 | luc2P | No | hPEST | minP + hypoxia RE | Hygro |
| pGL4.43 | luc2P | No | hPEST | minP + xenobiotic RE | Hygro |
| pGL4.44 | luc2P | No | hPEST | minP + AP1 RE | Hygro |
| pGL4.45 | luc2P | No | hPEST | minP + interferon stimulated RE | Hygro |
| pGL4.47 | luc2P | No | hPEST | minP + Sis-inducible element RE | Hygro |
| pGL4.48 | luc2P | No | hPEST | minP + SMAD3/SMAD4 binding element RE | Hygro |
| pGL4.49 | luc2P | No | hPEST | minP + TCF-LEF RE | Hygro |
| pGL4.50 | luc2 | No | No | CMV | Hygro |
| pGL4.51 | luc2 | No | No | CMV | Neo |
| pGL4.52 | luc2P | No | hPEST | minP + STAT5 RE | Hygro |
| pGL4.53 | luc2 | No | No | phosphoglycerate kinase (PGK) | No |
| pGL4.54 | luc2 | No | No | thymidine kinase (TK) | No |
| pGL4.70 | hRluc | Yes | No | No | No |
| pGL4.71 | hRlucP | Yes | hPEST | No | No |
| pGL4.72 | hRlucCP | Yes | CL1-hPEST | No | No |
| pGL4.73 | hRluc | No | No | SV40 | No |
| pGL4.74 | hRluc | No | No | HSV-TK | No |
| pGL4.75 | hRluc | No | No | CMV | No |
| pGL4.76 | hRluc | Yes | No | No | Hygro |
| pGL4.77 | hRlucP | Yes | hPEST | No | Hygro |
| pGL4.78 | hRlucCP | Yes | No | No | Hygro |
| pGL4.79 | hRluc | Yes | No | No | Neo |
| pGL4.80 | hRlucP | Yes | hPEST | No | Neo |
| pGL4.81 | hRlucCP | Yes | CL1-hPEST | No | Neo |
| pGL4.82 | hRluc | Yes | No | No | Puro |
| pGL4.83 | hRlucP | Yes | hPEST | No | Puro |
| pGL4.84 | hRlucCP | Yes | CL1-hPEST | No | Puro |
| Table 2. pNL Reporter Vectors. | |||||
| Vector | Reporter Gene | Multiple Cloning Region | Protein Degradation Sequence | Gene Promoter | Mammalian Selectable Marker |
|---|---|---|---|---|---|
| pNL1.1[Nluc] | Nluc | Yes | No | No | No |
| pNL1.1.CMV[Nluc/CMV] | Nluc | No | No | CMV | No |
| pNL1.2[NlucP] | NlucP | Yes | hPEST | No | No |
| pNL1.3[secNluc] | secNluc | Yes | No | No | No |
| pNL1.3.CMV [secNluc/CMV] | secNluc | No | No | CMV | No |
| pNL2.1[Nluc/Hygro] | Nluc | Yes | No | No | Hygro |
| pNL2.2[NlucP/Hygro] | NlucP | Yes | hPEST | No | Hygro |
| pNL2.3[secNluc/Hygro] | secNluc | Yes | No | No | Hygro |
| pNL3.1[Nluc/minP] | Nluc | Yes | No | minP | No |
| pNL3.2.CMV | NlucP | No | hPEST | CMV | No |
| pNL3.2.NF-κB-RE [NlucP/NF-κB-RE/ Hygro] | NlucP | No | hPEST | minP | Hygro |
| pNL3.2[NlucP/minP] | NlucP | Yes | hPEST | minP | No |
| pNL3.3[secNluc/minP] | secNluc | Yes | No | minP | No |
| pNLCoI1[luc2-P2A-NlucP/Hyg] | luc2, NlucP | Yes | Yes for NlucP, No for luc2 |
No | Hygro |
| pNLCoI2[luc2-P2A-Nluc/minP/Hyg] | luc2, NlucP | Yes | Yes for NlucP, No for luc2 |
minP | Hygro |
| pNLCoI3[luc2-P2A-NlucP/CMV/Hyg] | luc2, NlucP | No | Yes for NlucP, No for luc2 |
CMV | Hygro |
| pNLCoI4[luc2-P2A-NlucP/PGK/Hyg] | luc2, NlucP | No | Yes for NlucP, No for luc2 |
PGK | Hygro |
| Table 3. Luciferase Reporter Assays. | ||||
| Assay System | Gene Assayed | Single-Sample or Plate Assay | Signal Stability | Live-Cell Assay |
|---|---|---|---|---|
| Single Reporter | ||||
| Nano-Glo® Luciferase Assay System | Nluc, secNluc | Single or Plate2 | Long (≥2.0h) | Yes3 |
| Luciferase Assay System | luc, luc+, luc2 | Single or Plate1 | Short (<0.5h) | No |
| Steady-Glo® Luciferase Assay System | luc, luc+, luc2 | Plate2 | Long (>0.5h) | No |
| Bright-Glo™ Luciferase Assay System | luc, luc+, luc2 | Plate2 | Long (>0.5h) | No |
| ONE-Glo™ Luciferase Assay System | luc, luc+, luc2 | Plate2 | Long (≥45 minutes) | No |
| ONE-Glo™ EX Luciferase Assay System | luc, luc+, luc2 | Plate2 | Long (≥2 hours) | No |
| Renilla Luciferase Assay System | Rluc, hRluc | Single or Plate1 | Short (<0.5h) | No |
| Renilla-Glo® Luciferase Assay System | Rluc, hRluc | Plate2 | Long (≥60 minutes) | No |
| Dual Reporter | ||||
| Nano-Glo® Dual-Luciferase® Reporter Assay System | Nluc, luc, luc+, luc2 | Plate2 | Long (≥2h) | No |
| Dual-Glo® Luciferase Assay System | luc+, luc2, Rluc, hRluc | Plate2 | Long (>0.5h) | No |
| Dual-Luciferase® Reporter Assay System | luc+, luc2, Rluc, hRluc | Single (Cat.# E1910) | Short (<0.5h) | No |
|
|
|
Plate1 (Cat.# E1980) | Short (<0.5h) | No |
| Live-Cell | ||||
| Nano-Glo® Live Cell Assay System | Nluc | Plate | Short | Yes |
| Nano-Glo® Vivazine™ Live Cell Substrate | Nluc | Plate | Long | Yes |
| Nano-Glo® Endurazine™ Live Cell Substrate | Nluc | Plate | Long | Yes |
| EnduRen™ Live Cell Substrate | Rluc, hRluc | Plate1 | Long (>0.5h) | Yes |
| ViviRen™ Live Cell Substrate | Rluc, hRluc | Plate | Short (<0.5h) | Yes |
1Use with plates only when the luminometer has a reagent injector.
2We do not recommend the use of this product with automated reagent injectors.
3Use for live-cell assays by quantifying the secreted form of NanoLuc® luciferase in culture medium.