qPCR and RT-qPCR
A comprehensive introduction to qPCR and RT-qPCR methods.
This guide provides a comprehensive introduction to qPCR and RT-qPCR methods. For information on related products, visit the PCR product listing.
Introduction to qPCR and RT-qPCR
In the realm of molecular biology, the ability to accurately amplify and quantify genetic material is paramount for research and diagnostic purposes. Two advanced techniques that have revolutionized this field are quantitative PCR (qPCR) and Reverse Transcription Quantitative PCR (RT-qPCR). These methods provide precise, real-time data on DNA and RNA, respectively, enabling a wide array of applications from gene expression analysis to pathogen detection.
Quantitative Real-Time PCR (qPCR)
Quantitative Polymerase Chain Reaction (qPCR), also known as quantitative real-time PCR, is a laboratory technique used to amplify and simultaneously quantify a targeted DNA molecule in real time. Specially designed instruments amplify the target using thermal cycling and detect and monitor the fluorescence produced by PCR product accumulation. DNA-binding dyes, fluorescently labeled nucleic acid probes, and fluorescently labeled PCR primers can be used to detect the amplified product.
Why qPCR?
If you need to detect DNA sequences of low abundance in a sample or quantify the amount of DNA or gene expression levels in a sample, qPCR can precisely measure the PCR products. This technique, which measures the accumulation of PCR products as it occurs during amplification, can deliver a great deal of information about a sample in a short amount of time. While optimizing qPCR can seem overwhelming, consideration of a few important parameters will significantly increase your chances of success with qPCR.

What is the Difference Between Dye and Probe Detection for qPCR?
There are two common types of fluorescent reporters used in qPCR assays, double-stranded DNA binding dyes and labeled primers or probes. Choosing the type of reporter for your qPCR assay depends on the objectives of your experiment.
Dye-Based Detection for qPCR
Double-stranded DNA binding dyes bind to the double-stranded DNA product of PCR amplification. The unbound dye fluoresces at a much lower level than bound dye, therefore the level of fluorescence is directly proportional to the amount of amplified DNA produced. One advantage of using these dyes is that they will work with standard PCR primers, eliminating the need to acquire specially labeled primers or probes. However, the signal detected is not sequence-specific and you will need to perform additional analysis such as melt curve analysis to confirm that the correct sequence has been amplified.
Probe-Based Detection for qPCR
Probe-based qPCR relies on a reporter-quencher mechanism where sequence-specific probe that is labeled with a fluorescent reporter and a quencher molecule bind to the template. The fluorescent signal appears as probes specifically bind and interact with amplified target produced in the reaction. You will also need amplification primers for your target, but the probe-based detection means your results are more specific compared to the dye-based methos. One main advantage of using probe-based qPCR is that it provides the opportunity for multiplexing. By incorporating different sequence-specific probes labeled with different colored fluorescent reporter dyes, you can increase throughput by detecting several distinct sequences from the same qPCR.
Why use Melt Curve Analysis following qPCR?
You can verify that you have the correct qPCR amplification product by performing melt curve analysis. These curves are generated by allowing the PCR products to form double-stranded DNA at a lower temperature (approximately 60°C) and then slowly ramping the temperature to denaturing levels (approximately 95°C). As the DNA strands separate, the double-stranded DNA binding dye dissociates, and fluorescent signal is lost. The temperature at which 50% of double-stranded DNA (dsDNA) dissociates into single-stranded DNA, is known as the melt temperature (Tm). Both product length and sequence affect Tm, so the melt curve can be used to characterize amplicons present. Nonspecific amplification products can be identified by broad peaks in the melt curve or peaks with unexpected Tm values. By distinguishing specific and nonspecific amplification products, melt curve analysis adds a quality control aspect during routine use. The generation of melt curves is not possible with some probe-based detection methods that rely on the 5´→3´ exonuclease activity of Taq DNA polymerase, such as TaqMan® technology.
What are some common types of probes used for qPCR?
Some qPCR strategies employ complementary nucleic acid probes to quantify a DNA target, detect sequence polymorphism or measure copy number. Probes for qPCR contain a complementary sequence that allows the probe to anneal to the accumulating PCR product. There are many types of fluorescent labeled probes used in qPCR that differ in their configuration and how they produce fluorescent signal.
Hydrolysis probes: These probes are labeled with a fluorophore at the 5´-end and a quencher at the 3´-end. Because the two reporters are in close proximity, the fluorescent signal is quenched. During the annealing step, the probes hybridize to the PCR products generated in previous amplification cycles. The resulting probe:target hybrid is a substrate for the 5´→3´ exonuclease activity of the DNA polymerase, which degrades the annealed probe and liberates the fluor (1). The fluorophore, freed from the effects of the energy-absorbing quencher, produces a fluorescent signal, and as the amplification cycles continue, the accumulation of PCR product is monitored by the resulting increase in fluorescence.
Hairpin probes: These probes, also known as molecular beacons, contain inverted repeats separated by a sequence complementary to the amplification target. The repeats within the probe anneal to each other and form a hairpin structure, which brings the fluorophore at the 5´-end and a quencher at the 3´-end into close proximity. This results in little fluorescent signal. Hairpin probes are designed so that the probes bind preferentially to the amplification target’s sequence over retaining their hairpin structure. As the amplification reaction progresses, increasing amounts of the probe anneal to the accumulating PCR product. As a result, the fluorophore and quencher become physically separated. The fluorophore is no longer quenched, and the level of fluorescence increases. One advantage of this technique is that hairpin probes are less likely to mismatch than hydrolysis probes (2).
Reverse Transcription quantitative PCR (RT-qPCR)
RT-qPCR uses reverse transcription to convert RNA into a cDNA template prior to amplification. This method provides a means for sensitive and accurate quantitation of RNA levels.

RT-qPCR: 1-step vs. 2-step
RT-qPCR can be done in a single step or by dividing that process into two steps. The 1-step method saves time and work by combining the reverse transcription and amplification reactions, but you can’t save the cDNA to analyze later. In the 2-step process, you perform the reverse transcription reaction first, and then use the cDNA product in a separate amplification reaction. With the 2-step process, you have the option to save the cDNA produced during the reverse transcription reaction to use later, making it possible to analyze different target sequences within an amplified sample.

|
1-Step |
2-Step |
||
| Select 1-Step if: | Select 2-Step if: | ||
|
You want to save time |
You prefer to keep the cDNA and use it for multiple reactions | ||
|
You have only a few targets |
You have a limited amount of sample | ||
|
You want high throughput or automation |
You are assaying many targets per sample | ||
|
Advantages |
Disadvantages | Advantages | Disadvantages |
|
Reduced chance of cross-contamination during the procedure |
Less able to troubleshoot or optimize | Optimized performance of both RT and PCR steps | |
|
Less variation, highly reproducible |
Can be less sensitive | cDNA can be used for multiple reactions | Less convenient, more time consuming |
|
Faster results |
Can be more sensitive | Higher risk of contamination (compared to 1-step method) | |
Although qPCR and RT-qPCR can seem daunting to learn, these techniques are powerful tools for measuring gene expression and other genomic analysis applications. Choosing the type of fluorescent reporter for your qPCR assay is a good starting point. Using dye-based assays are more cost effective, while probe-based assays make it possible to multiplex. RT-qPCR is used to quantify RNA. RT-qPCR can be performed in a single step to save time or divided into two steps. Two step RT-qPCR allows cDNA produced during reverse transcription to be set aside for amplification later.
qPCR and RT-qPCR Products
GoTaq® qPCR and RT-qPCR kits are available for dye-based or probe-based real-time PCR approaches. GoTaq® qPCR Systems contain BRYT Green Dye, which provides maximum amplification efficiency and greater fluorescence than SYBR Green.
GoTaq® Probe Systems are ready-to-use master mixes that simplify reaction assembly for hydrolysis probe-based detection.
Frequently Asked Questions (FAQ)
What is qPCR, also referred to as qRT-PCR?
Quantitative polymerase chain reaction (qPCR) or quantitative real time PCR (qRT-PCR), is a method for measuring the amount of DNA that is present in a sample. Performing PCR in real time means that product formation is measured at each cycle as it is occurring by measuring a change in a fluorescent reporter signal. With traditional, end-point PCR the signal is measured only once after the cycling program has ended. qPCR, sometimes referred to as quantitative real-time PCR or qRT-PCR, should not be confused with RT-qPCR, which is reverse transcription qPCR, where the starting template is RNA that is reverse transcribed into a cDNA target prior to qPCR.
What kind of reporter can I use in my qPCR assay?
There are two general types of fluorescent reporters that are used in qPCR assays:
Double-stranded DNA-binding Dyes
Double-stranded DNA-binding dyes are dyes that bind to the minor groove of double-stranded DNA (dsDNA). The free dye has a very low fluorescence, whereas the bound dye has a high fluorescence. The more double-stranded PCR product that is produced, the greater the fluorescence signal will be. BRYT Green® Dye is an example of a dsDNA-binding dye. These dyes allow you to use standard PCR primers, so there is no need to order special labeled primers. The disadvantage is that the signal is not sequence-specific, so you will need to verify that the reaction is producing the desired product.
Labeled Primer or Probe
The second type of reporter method uses a labeled primer or probe to detect the amplification product. There are many different ways to modify reporter signal generation. Real-time PCR using labeled oligonucleotide primers or probes employs two different fluorescent reporters and relies on energy transfer from one reporter (the energy donor) to a second reporter (the energy acceptor) when the reporters are in close proximity. The second reporter can be either a quencher or a fluor. If the second reporter is a quencher, the energy from the first reporter is absorbed but re-emitted as heat rather than light, leading to a decrease in fluorescent signal. Alternatively, if the second reporter is a fluor, the energy can be absorbed and re-emitted at another wavelength through fluorescent resonance energy transfer (FRET) and the progress of the reaction can be monitored by the decrease in fluorescence of the energy donor or the increase in fluorescence of the energy acceptor. Some qPCR strategies employ complementary nucleic acid probes to quantify the DNA target. These probes also can be used to detect single nucleotide polymorphisms (3,4). There are many different types of real-time PCR probes, including hydrolysis, hairpin and simple hybridization probes. Probes contain a complementary sequence that allows the probe to anneal to the accumulating PCR product but can differ in the number and location of the fluorescent reporters. TaqMan® Assays are an example of a method that uses a labeled probe. The big advantage that label-based methods offer is the ability to multiplex two or more targets in a single reaction by using different fluorescent reporters. This would allow you to measure an experimental gene and a control gene for normalization in the same reaction.
What is a qPCR amplification curve?
The primary output from your real-time PCR instrument is an amplification curve. The curve shows the normalized fluorescent signal versus the cycle number and thus shows the accumulation of amplified product. The amplification curve can be broken down into three parts. The baseline is the point early in the amplification process where very little product has been produced and the signal is still below the background noise of the assay. The exponential (log) phase occurs around the time that the reaction reaches the amplification threshold; and indicates that the product is doubling with every cycle. This phase can be seen more clearly if the results are graphed on a semi-log plot. This is the most critical phase for accurate quantification because it reflects the true amplification efficiency. The cycle threshold (Ct) value is determined in this phase, which is the point at which the fluorescence signal exceeds a predefined threshold level above the baseline noise. The third part of the amplification curve is the plateau phase, where amplification efficiency drops and fluorescent signal levels off indicating that no new product is being produced.

Source: BioRender
What is the amplification threshold?
The amplification threshold is calculated based on the background fluorescence over the baseline region of the amplification curves. The threshold represents the point where the reporter signal is significantly higher than the background levels.
The amplification threshold is a set fluorescence value that the PCR curve must cross. To find the cycle threshold (Ct), the software determines when the fluorescence signal rises above this threshold. There is no specific single equation for the threshold itself, as it is user-defined or software-defined based on the baseline noise of the reaction.
How do I use my results to quantitate my starting material?
Quantifying the amount of starting material is done using the quantification cycle (Cq; also known as Ct). The Cq is the cycle number where the amplification curve crosses the amplification threshold. Once you have the Cq value, there are two approaches for converting that value to template quantity. In absolute quantification, the Cq value can be used to determine the amount of an unknown sample using a standard curve derived from reactions which contain known amounts of the target. The standard curve is generated by plotting the Cq versus concentration (log concentration) for the standard curve samples. The concentration of unknown samples then can be determined using an equation derived from the linear regression analysis of the standard curve.
Most real-time PCR instrument software can also perform relative quantification using the Cq values. Relative quantification is based on the premise that each template is replicated with each cycle; therefore, n cycles would produce 2n product. This relationship also works in reverse; for example, two samples with a Cq difference of 1 would have a twofold difference in starting template amount. This is the basis for the simplest algorithm for relative quantitation (5); however, this is only accurate if amplification efficiency is 100 percent. Amplification efficiency can be calculated from a standard curve (done during assay validation) using the equation shown in Figure 1 (6). If amplification efficiency is less than 100%, then relative quantification calculations must be adjusted to correct for the decreased efficiency (7).
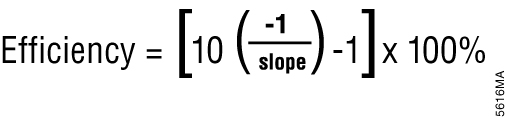
Figure 1. Equation for determining amplification efficiency.
How can I ensure success when doing RT-qPCR?
RT-qPCR is the same as qPCR but using a starting template of RNA that is reverse transcribed (RT) to cDNA prior to amplification. A successful RT-qPCR assay is dependent on the success of the RT reaction, and successful reverse transcription depends on RNA integrity and purity. There are several methods for evaluating your RNA before proceeding to the RT step. RNA integrity can be assessed qualitatively by gel electrophoresis or quantitatively using systems such as the Agilent Bioanalyzer, which uses microfluidics to size‐separate and quantitate RNA. RNA quantity and purity can be assessed by absorbance. The quantity is determined by absorbance at A260, and quality is measured using a ratio of absorbance at A260/A280. High‐quality RNA should have a minimum A260/280 ratio of about 2. Spiking experiments, in which RNA of unknown quality is added to a control assay with template also commonly referred to as an internal amplification control or internal positive control assay, example IAC RT-qPCR Control cat.# AM2040, can be used to detect inhibitors. It is a good idea to include a no-RT qPCR control reaction to detect gDNA contamination. Finally, quantifying the RNA will help normalize the amount of RNA you are including in each reaction.
Anytime you are working with RNA, RNA degradation is a concern. We recommend that you have a dedicated, ribonuclease-free (RNase-free), area for all RNA work. Procedures for creating and maintaining a RNase-free environment to minimize RNA degradation should be followed faithfully. Using an RNase inhibitor (e.g., Recombinant RNasin® Ribonuclease Inhibitor, Cat. #N2111) is strongly recommended.
What is the difference between 1-step and 2-step RT-qPCR?
The fundamental difference between these two approaches is whether the RT step is performed separately from the PCR or in the same tube. For one-step RT-PCR, the RT and PCR steps are performed sequentially in the same tube using the entire amount of the cDNA synthesis products as the template for PCR. For two-step RT-PCR, the RT and PCR steps are also performed sequentially, but only a portion of the cDNA products is used as a template for PCR, which is performed in a separate tube. For this reason, one-step RT-qPCR is is ideal when only one or a few targets is being measured and sample is not limited. However, two-step RT-PCR allows multiple PCRs from a single RT reaction, which works well for quantifying multiple targets or for doing replicate assays.
One-step RT-PCR commonly uses gene-specific primers for both the RT and PCR steps, with one of the PCR primers also acting as the RT primer. Two-step RT-qPCR can use oligo(dT) primers or random primers for the RT step.
What is Absolute Quantitation in qPCR?
Absolute quantitation in qPCR refers to the precise determination of the absolute number of target DNA copies in a sample. This method requires the construction of a standard curve from serial dilutions of a known concentration of target DNA. The amount of target DNA in unknown samples is then interpolated from this standard curve, providing an exact number of copies.
What is Relative Quantitation in qPCR?
Relative quantitation measures the change in expression of a target gene relative to a reference gene or control sample. This method is commonly used to compare gene expression levels across different samples or conditions, providing insights into gene regulation and expression patterns.
What are the Calculation Methods for Relative Quantitation?
There are two primary methods for calculating relative quantitation in qPCR:
- Standard Curve Method for Relative Quantitation
- Comparative Cq Method for Relative Quantitation
Standard Curve Method for Relative Quantitation
The standard curve method involves generating a standard curve from serial dilutions of a reference sample. The target gene and reference gene are quantified in the same sample, and their relative quantities are calculated based on the standard curves. This method provides accurate quantitation but can be labor-intensive due to the need for standard curves for each gene.
Comparative Cq Method for Relative Quantitation (ΔΔCq Method)
The Comparative Cq method, also known as the ΔΔCq method, is a simpler and more widely used approach. This method calculates the difference in the threshold cycle (Cq) values between the target gene and the reference gene within each sample. The relative expression level is then determined by comparing these differences across different samples or conditions. This method is less labor-intensive as it does not require the generation of standard curves for each gene.
When to Use Each Method?
- Absolute Quantitation: Use this method when you need precise quantification of the target DNA copy number. It is essential in applications such as viral load determination, gene copy number variation studies and absolute quantification of gene expression levels.
- Relative Quantitation: This method is ideal for comparing gene expression levels across different samples or experimental conditions. It is commonly used in studies involving gene expression profiling, validation of microarray data, and functional genomics research.
- Standard Curve Method for Relative Quantitation: Use this method when high accuracy in relative quantitation is required, especially when dealing with multiple genes that need to be compared within the same experiment.
- Comparative Cq Method for Relative Quantitation: This method is suitable for high-throughput experiments where multiple samples are analyzed, and a simpler, faster approach is needed. It is widely used due to its ease of use and minimal requirement for standard curve preparation.
Standard Curve Method for Absolute Quantitation
The standard curve method for absolute quantitation involves the creation of a standard curve from a series of known concentrations of the target DNA. Each sample's Cq value is plotted against the logarithm of the known DNA concentrations to generate the curve. The unknown sample's DNA concentration is then determined by interpolation from this standard curve, providing an exact copy number of the target DNA.
Overcoming Inhibitors in qPCR
PCR inhibitors can significantly disrupt qPCR experiments, causing underestimation of nucleic acid targets or amplification failure. These inhibitors may originate from sample materials, co-purify with the DNA/RNA template, or arise during reaction setup. Detecting and mitigating their effects is crucial for successful qPCR. Key Indicators of Inhibition:
- Changes in Cq Values: An elevated quantification cycle (Cq) value may suggest inhibition but could also reflect reduced target amounts. Distinguishing these requires internal PCR controls (IPC) or template dilution tests.
- Slope of Standard Curves: A standard curve slope outside the range of -3.3 to -3.6 indicates poor amplification efficiency and potential inhibition.
- Amplification Curve Morphology: Alterations in curve shape, such as flattening or failure to cross the detection threshold, often signal inhibition.
Common Types of Inhibition:
- Polymerase Inhibition: Often visible as a delayed or altered amplification curve.
- Reverse Transcriptase Inhibition: Manifests as shifts in Cq values, affecting target RNA quantification.
- Signal Interference: Inhibitors can interfere with fluorescence dyes or probes, reducing signal strength.
Strategies for Detection and Mitigation:
- Include IPCs to identify inhibition without template quantity concerns.
- Perform serial dilutions to evaluate Cq consistency and efficiency.
- Optimize sample preparation to minimize carryover of inhibitory substances.
Inhibitor Resistant Products
GoTaq® Endure polymerase is engineered for robust performance, providing reliable amplification even in the presence of common PCR inhibitors, ensuring consistent and accurate results with challenging samples.
qPCR Terms
ΔR: baseline-corrected fluorescence (R) — R minus average R over baseline region. This value represents the net increase in fluorescence signal attributable to the specific amplification of the target sequence. As the target DNA is amplified during the PCR cycles, the ΔRn value increases. ΔRn is used to normalize fluorescent data and determine Cq values.
Passive reference: free dye (unassociated with amplification product, often ROX or CXR dyes) used for normalization to control for experimental and optical variation).
Rn: normalized fluorescence — R divided by passive reference.
ΔRn: baseline-corrected, normalized fluorescence.
Experimental Target: the target gene, transcript, channel or dye of interest.
Control Target: normalized, endogenous control, reference gene, transcript, channel, dye or housekeeping gene.
Experimental Sample: unknown or treated sample.
Control Sample: calibrator, reference sample.
RT-qPCR Resources
General
Gene Quantification: www.gene-quantification.de
A–Z of quantitative PCR (Editor: S.A. Bustin ), International University Line (IUL), La Jolla, CA, USA.
Design, Validation, Controls, Normalization
Sean Taylor, et al. (2011) A MIQE Case Study — Effect of RNA Sample Quality and Reference Gene Stability on Gene Expression Data Bio-Rad tech note 6245.
Garbarino-Pico, E. et al. (2007) Immediate early response of the circadian polyA ribonuclease nocturnin to two extracellular stimuli. RNA 13, 745–755. PMID:17400819.
Pre Designed qPCR Primers
Primer Bank
RTPrimerDB
Primer Design Software
Primer3
Primer-BLAST
Overview of the MIQE Guidelines for RT-qPCR
What are the MIQE Guidelines?
The MIQE (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) guidelines are a set of standards developed to ensure the reliability, reproducibility, and transparency of quantitative real-time PCR (qPCR) experiments. Published in 2009 by Bustin et al., the MIQE guidelines provide a comprehensive framework for designing, conducting, and reporting qPCR experiments. The guidelines cover all aspects of the experimental process, from sample preparation to data analysis, aiming to improve the quality and consistency of qPCR results across different laboratories and studies.
Key Components of the MIQE Guidelines
The MIQE guidelines encompass several key components that researchers must consider when performing RT-qPCR experiments:
- Experimental Design:
- Clear definition of experimental aims and hypotheses.
- Proper control of variables and replication to ensure statistical validity.
- Include appropriate negative and positive controls.
- Sample Quality and Preparation:
- Assessment of RNA quality and integrity.
- Use of appropriate methods for RNA extraction and quantification.
- Minimize contamination and degradation during sample handling.
- Reverse Transcription:
- Selection of suitable reverse transcriptase enzymes and priming strategies.
- Verification of reverse transcription efficiency and absence of genomic DNA contamination.
- Primer and Probe Design:
- Validation of primer and probe specificity and efficiency.
- Avoidance of secondary structures and primer-dimer formation.
- qPCR Protocol:
- Optimization of reaction conditions, including reagent concentrations and thermal cycling parameters.
- Use of appropriate controls, such as no-template controls and reverse transcription minus controls.
- Data Analysis:
- Accurate determination of Cq (quantification cycle) values.
- Application of proper normalization strategies using reference genes.
- Use of appropriate statistical methods for data analysis and interpretation.
- Reporting and Documentation:
- Comprehensive reporting of experimental details, including reagent information, instrument settings, and data analysis methods.
- Availability of raw data and analysis protocols for peer review and reproducibility.
Download Interactive MIQE Checklist
Related Resources
- Guidelines for Successful qPCR Master Mix Comparison
- Download Interactive MIQE Checklist link to the interactive MIQE Guidelines
- Guidelines for a Comparison of Reagent Performance
- qPCR: Guidelines for Validating a Multiplex Assay
- Primer selection guide for use with GoScript® Reverse Transcription Mixes link
WEBINARS
Related Products
Categories
Related Groups
References
- Holland, P.M. et al. (1991) Detection of specific polymerase chain reaction product by utilizing the 5′→3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc. Natl. Acad. Sci. USA 88, 7276–80.
- Tyagi, S. et al. (1998) Multicolor molecular beacons for allele discrimination. Nat. Biotechnol. 16, 49–53.
- Lee, L.G., Connell, C.R. and Bloch, W. (1993) Allelic discrimination by nick-translation PCR with fluorogenic probes. Nucleic Acids Res. 21, 3761–6.
- Bernard, P.S., Lay, M.J. and Wittwer, C.T. (1998) Integrated amplification and detection of the C677T point mutation in the methylenetetrahydrofolate reductase gene by fluorescence resonance energy transfer and probe melting curves. Anal. Biochem. 255, 101–7.
- Livak, K.J. and Scmittgen, T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–8.
- Bustin, S.A. (2004) A–Z of quantitative PCR. International University Line, La Jolla.
- Pfaffl, M.W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45.
For Research use Only. Not for Use in Diagnostic Procedures.