Using GoTaq® Long PCR Master Mix for T-Vector Cloning
Alisha Truman, Brad Hook and Doug Wieczorek Promega Corporation
Publication Date: June, 2013; tpub 120
Abstract
The pGEM®-T Easy Vector is a convenient system to easily clone PCR products. The vector contains 3´-terminal thymidines to improve ligation efficiency of PCR products generated with A-tails by preventing recircularization of the vector and providing compatible overhangs for the ligation of the PCR product. Thermostable polymerases such as GoTaq®/Taq/AmpliTaq®, Tfl and Tth are known to successfully ligate into the pGEM®-T Easy Vector System. Here we show that PCR fragments generated with the GoTaq® Long PCR Master Mix can be successfully ligated into the pGEM®-T Easy Vector.
Introduction
The pGEM®-T Easy Vector is a linearized vector with 3´-terminal thymidines at both ends. These T-overhangs at the insertion site improve ligation efficiency of PCR products generated from thermostable polymerases that generate 3´ A-tailed fragments such as GoTaq®/Taq/AmpliTaq®, Tfl and Tth. These enzymes generate an A-tailed fragment as a result of their 5´→ 3´ exonuclease activity and lack of 3´→5´ exonuclease activity. In contrast, proofreading DNA polymerases such as Vent and Pfu generate blunt-ended fragments. These blunt-ended fragments require modification by an A-tailing procedure before ligation into a T vector.
The pGEM®-T Easy Vector allows blue/white colony screening to detect colonies with and without an insert. Ligation of an insert into the plasmid disrupts the coding for β-galactosidase, resulting in white colonies. Blue colonies contain the intact plasmid (no insert).
GoTaq® Long PCR Master Mix utilizes recombinant Taq DNA polymerase as well as a small amount of a recombinant proofreading DNA polymerase. This 3´→5´ exonuclease activity (proof reading) enables amplification of long targets. Here we show that despite the presence of a small amount of 3´→5´ exonuclease activity, the GoTaq® Long PCR Master Mix generates PCR products that can be successfully ligated into the pGEM®-T Easy Vector System.
Methods
Amplification and Purification
We used the pGL4.10[luc2] Vector (Cat.# E6651) to amplify the luciferase reporter gene luc2 (1,652bp) using the forward primer: 5´-TGGTGCGATCGCCATGGAAGATGCCAAAAACATTAAGAAGGGCCCAG-3´, the reverse primer: 5´-GTGAGTTTAAACCACGGCGATCTTGCCGCCCT-3´ and three different enzyme mixes:
- GoTaq® Long PCR Master Mix (Cat.# M4021)
- GoTaq® Green Master Mix (Cat.# M7122)
- PfuUltra Hotstart DNA Polymerase (Agilent Cat.# 600390)
The GoTaq® Green Master Mix is expected to generate A-tailed PCR fragments that can be cloned into the pGEM®-T Easy Vector. PfuUltra is expected to remove A-overhangs from the PCR product due to its proofreading ability.
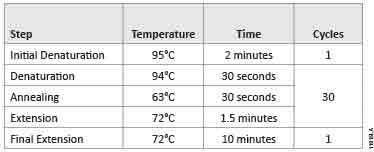
We amplified luc2 using GoTaq® Long PCR Master Mix according to the protocol (TM359) with the suggested parameters (Table 1) on an MJ Research PTC-200 Thermal Cycler.
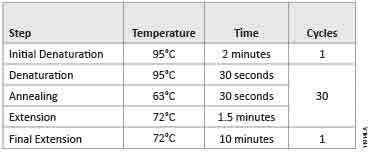
We also amplified the luc2 gene using the PfuUltra Hotstart DNA Polymerase and GoTaq® Green Master Mix as described in the PfuUltra Hotstart DNA Polymerase instruction manual. Both reactions were amplified on the Bio-Rad DNA Engine® Thermal Cycler (Table 2).
uFollowing amplification, we pooled three reactions from each enzyme mix for further analysis. The pooled PCR products were run on a 0.8% agarose gel and DNA was purified from gel slices using the Wizard® SV Gel and PCR Clean-up System (Cat.# A9281), and the concentration and purity of the cleaned-up PCR products were determined by NanoDrop®-1000 spectrophotometry. Finally, we visualized the purified products on a 0.8% agarose gel.
Ligation and Transformation
We ligated with the amplified luc2 DNA generated from PfuUltra Hot Start DNA Polymerase, GoTaq® Green Master Mix, and GoTaq® Long PCR Master Mix reactions into the pGEM®-T Easy Vector using a 3:1 insert to vector ratios according to the protocol (#TM042). The resulting pGEM®-T-luc2 clones were transformed into JM109 High Efficiency Competent Cells (>108cfu/µg; Cat.# L2001). Following transformation, the cells (100µl) were plated in triplicate onto LB plates with ampicillin/IPTG/X-Gal and incubated for 18 hours at 37°C. We inoculated liquid cultures with single white colonies and grew them overnight in LB with ampicillin. We used the PureYield™ Plasmid Miniprep System (Cat.# A1223) to isolate the plasmid DNA. To confirm the expected size of the DNA insert, we digested the purified plasmid DNA with EcoRI (Cat.# R6011) according to the Restriction Enzyme Protocol Technical Manual #TM367 and visualized the products on a 0.8% agarose gel.
Competent E. coli Comparison
To compare the efficiency between competent cell types, we transformed both the Pro 5-alpha Competent Cells (>109cfu/µg; Cat.# L1221), JM109 Competent Cells (>107cfu/µg) and JM109 High Efficiency Competent Cells (>108cfu/µg) with ligation reactions from the GoTaq® Long PCR reaction. Fifty microliters of each competent cell type was transformed according to the pGEM®-T Easy Vector System Technical Manual #TM042 and added to 950µl of SOC medium. We plated 100µl of each transformation mix (10% of total) in duplicate onto LB plates with ampicillin/IPTG/X-Gal, and used blue/white screening to determine colony counts.
Results
Amplification and Purification
Following purification with the Wizard® SV Gel and PCR Clean-Up System, we determined the DNA concentration of the amplified DNA using a NanoDrop®-1000. All the amplified products had concentrations ≥22.1ng/µl and A260/A280 and A260/A230 values ≥1.90 (Table 3). We used agarose gel (0.8%) analysis to confirm the expected size of the amplified luc2 (1,652bp) product (Figure 1). DNA fragments generated from GoTaq® PCR Master Mix, GoTaq® Long PCR Master Mix, and PfuUltra Hot Start DNA Polymerase all showed a prominent fragment of DNA at ~1,600bp.
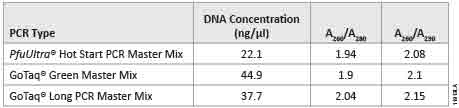
Table 3. DNA concentrations and purity ratios of the PCR products. PCR products were generated by GoTaq® Green Master Mix, GoTaq® Long PCR Master Mix, and PfuUltra Hot Start DNA Polymerase after Wizard® SV Gel and PCR Clean-Up.
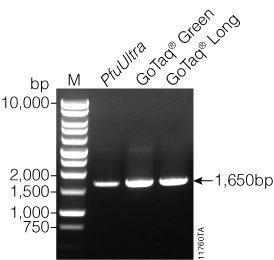
Figure 1. Gel analysis of purified PCR products. Five microliters of purified PCR product from GoTaq® PCR Master Mix (GoTaq® Green), GoTaq® Long PCR Master Mix (GoTaq® Long), and PfuUltra Hot Start DNA Polymerase (PfuUltra) on a 0.8% agarose gel. The expected size of the amplified luc2 fragment is 1,652bp. A prominent fragment at ~1,600bp was visible for PCR products from all three PCR reactions. Lane M: BenchTop 1kb Ladder (Cat.# G7541).
Ligation and Transformation
Inserts generated by GoTaq® Green Master Mix and GoTaq® Long PCR Master Mix resulted in blue/white colony counts of 61% and 77% white colonies, respectively. These results were comparable to the positive control plates (Table 4). Ligations with the insert generated by PfuUltra Hotstart DNA Polymerase resulted in one white colony between three plates, which corresponded to 3% white colonies. This indicates that most colonies did not contain vector with insert DNA.
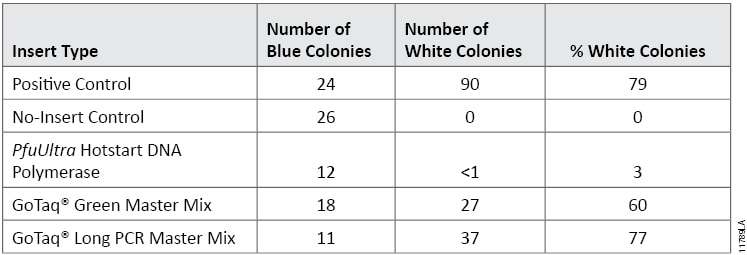
Table 4. Transformation Results. Blue and white colony counts and percent white colonies are shown for transformation reactions of the luc2 gene amplified using the PfuUltra Hotstart DNA Polymerase, GoTaq® Green Master Mix or GoTaq® Long PCR Master Mix and ligated into pGEM®-T Easy Vector. Vectors ligated with control DNA provided in the pGEM®-T Easy Vector System were used as the positive control, and vector-only transformations were used as negative controls. Values are averages from three plates.
We used an EcoRI restriction enzyme digest to confirm the size of the insert. EcoRI restriction sites are located on each end of the pGEM®-T Easy Vector cloning site. Plasmids generated by ligation of PCR products from each of the three polymerase reactions revealed fragments of approximately 3,000bp after digestion, indicative of the pGEM®-T Easy Vector (Figure 2). Vectors ligated with GoTaq® Green Master Mix PCR product or GoTaq® Long PCR Master Mix PCR products also resulted in fragments of approximately 1,650bp as expected for the luc2 insert. The single white colony generated from vectors ligated with PfuUltra Hotstart DNA Polymerase PCR product resulted in vector only and no visible fragment for the insert DNA.
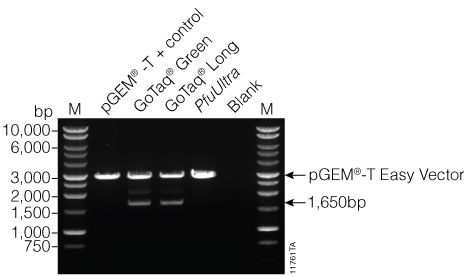
Figure 2. Restriction digestion analysis of the pGEM®-T-luc2 clones. Five microliters of each EcoRI digestion of plasmid from white colonies to confirm the size of insert on a 0.8% agarose gel. The pGEM®-T Easy Vector is present for all samples at 3,000bp as expected. Digests of plasmids ligated with GoTaq® Green Master Mix (GoTaq® Green) or GoTaq® Long PCR Master Mix (GoTaq® Long) PCR products indicate a 1,650 bp fragment for the luc2 insert. Digestion of plasmid purified from the single white colony from the PfuUltra Hotstart DNA Polymerase PCR product (PfuUltra) plates resulted in a vector-only fragment with no visible insert. BenchTop 1kb Ladder was used as a marker (M; Cat.# G7541).
Competent E. coli Comparison
To compare Pro 5-alpha Competent Cells, JM109 Competent Cells and JM109 High Efficiency Competent Cells a single ligation mix was transformed into each strain and a blue/white screen performed. All three competent cell types resulted in a similar percentage of white colonies (Table 5). Pro 5-alpha Competent Cells had significantly higher colony counts than the JM109 Competent Cells and estimates were made. In addition, the Pro 5-alpha Competent Cells colonies resulted in smaller colonies than the JM109 Competent Cells (Figure 3).
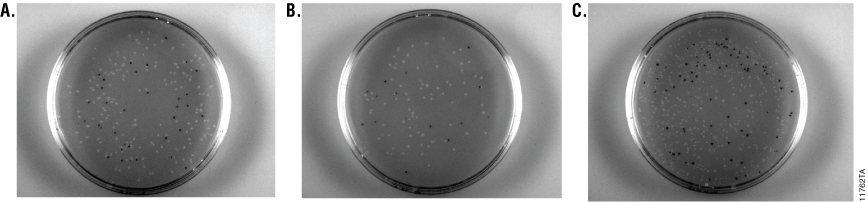
Figure 3. Overnight blue/white selection plates of different E. coli preparations transformed with the same ligation mix. JM109 Competent Cells (>107cfu/µg; Panel A), JM109 High Efficiency Competent Cells (>108cfu/µg; Panel B) and Pro 5-alpha Competent Cells (>109cfu/µg; Panel C) were transformed with pGEM®-T-luc2 construct. One hundred microliters of each bacterial transformation mix was plated on LB/Amp/IPTG/X-Gal plates and incubated overnight. Colonies containing the pGEM®-T-luc2 construct were identified using blue/white screening.
Summary
The GoTaq® Long PCR Master Mix successfully generated DNA fragments that could be ligated into pGEM®-T Easy Vector System without an A-tailing procedure and with ligation efficiencies similar to those observed with the GoTaq® Green Master Mix. PCR products generated with the GoTaq® Long PCR Master Mix and ligated with the pGEM®-T Easy Vector System can be transformed into either Pro 5-alpha Competent Cells, JM109 Competent Cells, or JM109 High Efficiency Competent Cells with similar blue/white colony percentages.