Establishing a Luminescent Biosensor in E. coli: The Modulated Acetosyringone Receptor Sensing System
F. Haenisch*, J. Aretz*, E. Brombacher*, N. Kessler*, N. C. Lübke*, J. Marschall*, A. Neshat*, S. Unthan*, F. Walter*, T. Wolf*, C. Rückert, J. Kalinowski and K. Niehaus
*These authors contributed equally to this work. Center for Biotechnology, Bielefeld University, Bielefeld, Germany
Publication Date: 2011
Abstract
In search for a cheap and reliable “hotness”-biosensor, we established a luminescent signaling pathway in E. coli. The receptor and signaling pathway of A. tumefaciens was transferred into E. coli Top 10 cells and connected to a luminescent readout. The signaling pathway allows for reliable quantification of the inducing molecule acetosyringone, a close relative of capsaicine, the hotness molecule. This project was part of the IGEM 2010 competition.
[Editor's Note: The International Genetically Engineered Machine competition (iGEM) is the premiere undergraduate Synthetic Biology competition. At the beginning of the summer, student teams are given a kit of biological parts from the Registry of Standard Biological Parts. They spend the summer working with their teams at their schools to build biological systems and operate them in living cells.]
Introduction
The model organism Agrobacterium tumefaciens is a soil bacterium and the causative agent of the crown gall disease in dicotyledonous plant species(1). The VirA receptor of A. tumefaciens recognizes phenols and initiates a signal transduction cascade for the expression of virulence (vir) genes. The initiation of an infection is controlled by a two-component system comprised of the transmembrane-bound sensor (VirA) and the intracellular response regulator (VirG;(1)(2)). Upon activation by phenol 3,5-dimethoxyacetophenone (acetosyringone), the histidine kinase VirA is autophosphorylated at its His-474 residue(3); the phosphor is then transferred to VirG(4)(5)(6)(Figure 1). VirG acts as a transcription factor and binds to a promoter containing the virulence box (vir Box); for example, the virB promoter(4)(7).
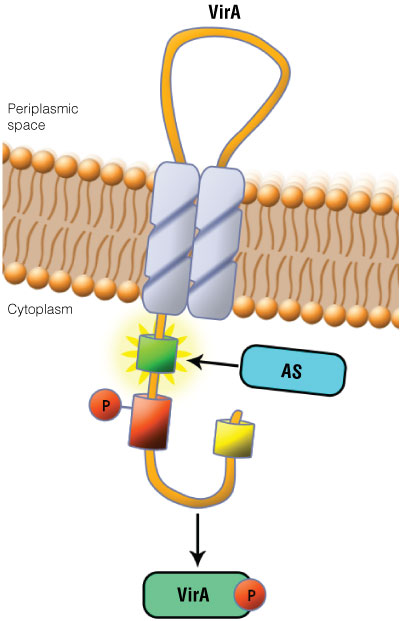
Figure 1. Schematic showing the VirA receptor location. In A. tumefaciens, the VirA receptor is located in the inner membrane with a loop structure in the periplasmatic space. The ligand acetosyringone (AS) activates VirA. After autophosphorylation of VirA, the phosphate is transferred to the response regulator VirG.
Expression System and BioBricks
The aim of our project was to transfer the very specific recognition and signaling mechanisms of A. tumefaciens into a standard expression system. We subcloned virA and virG into the E. coli TOP10 cells. To exclude a coactivation of other A. tumefaciens pathways, we also subcloned the inducible promoter virB. All the plasmid constructs we generated were conform using the guidelines of the BioBrick standard, and thus, can be assembled easily in any chosen order.
Readout System
We built a BioBrick with a luminescent readout system to quantify the target substance. For this purpose, the virG-inducible and vir box-containing promoter virB was cloned upstream of the firefly luciferase gene.
To design a new biosensor based on the receptor binding site of VirA, a proof of principle had to be established. We used the native ligand of VirA, the phenol acetosyringone, to induce a luciferase-based light signal in E. coli (Figure 2).
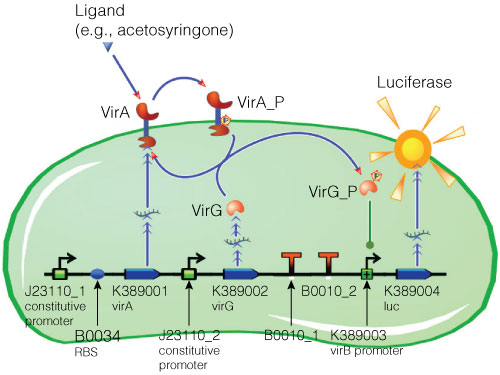
Figure 2. Model of acetosyringone-inducible luciferase expression in E. coli. Construct contains constitutive expression promoters for the two-component receptor system (virA + virG), the two-component system as well as the luciferase gene under control of the inducible virB promoter. Ligand binding to the VirA receptor initiates the internal signal transduction cascade through VirG phosphorylation. Activated transcription factor VirG binds to the vir Box-containing promoter virB. Luciferase expression occurs relative to the initial VirA ligand concentration. Gene numbers describe the database reference number of the BioBrick database.
BioBrick Assembly - Suffix Insertion
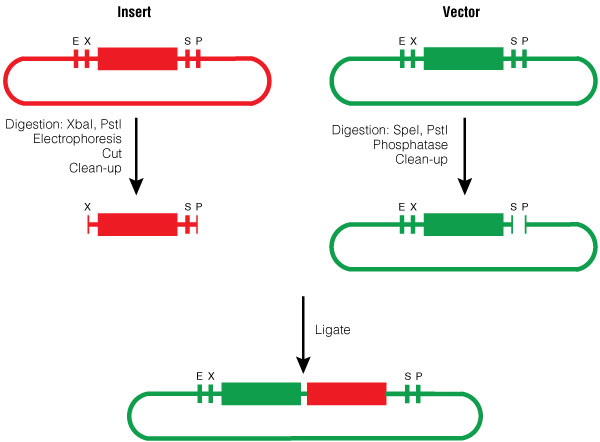
The insert was digested with XbaI and PstI and the pGL4.10[luc2] vector was digested with SpeI and PstI according to standard protocols. Following digestion, the vector was phosphorylated using shrimp alkaline phosphatase, cleaned with a PCR clean-up kit, and then ligated with the BioBrick insert (Figure 3).
Cultivation for Measuring mRFP and Luciferase Expression
E. coli Top 10 glycerol stocks were transformed with the pGL4.10[luc2]-BioBrick construct and cultivated with appropriate antibiotics at 37°C with shaking at 175 rpm. The handling of the bacterial culture depends on the readout strategy that will be used.
Measuring of Luciferase
For luciferase detection, we used the Promega Luciferase Assay System according to the manufacturer’s protocol. Bacterial cells were lysed in a high salt buffer (1M K2HPO4, 20mM EDTA, pH 7.8). Ninety microliters of the sample was exposed to one freeze-thaw cycle at –80°C. Following the freeze-thaw cycle, 300µl of freshly prepared lysis mix (1X Cell Culture Lysis Reagent, 1.25mg/ml lysozyme, 2.5mg/ml BSA) was added and mixed. The luminescent signal was detected using the Promega GloMax®-Multi Detection System with dual injector. (Injection volume of Luciferase Assay Reagent: 100µl, delay: 20 seconds, integration: 3 seconds).
Results
We established the complete VirA/G signaling system from A. tumefaciens in E. coli. A basal expression of luciferase without any acetosyringone induction could be obtained. After adding 50µM and 150µM of acetosyringone, we measured and characterized an induction signal using the reporter genes mRFP and luciferase. Induction with acetosyringone resulted in an increase in luciferase expression levels (compared to the basal expression; Figure 4).
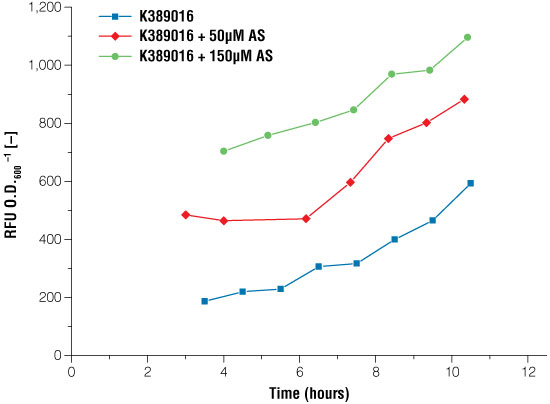
Figure 4. Signal intensity of luciferase readout system in E. coli under certain conditions of acetosyringone induction. Blue line: no induction, the basal expression of the luciferase can be seen; red line: induction with 50µM acetosyringone; green line: induction with 150µM acetosyringone. Measurements took place after growing the cells in shaking flasks.
To characterize the luciferase expression response to acetosyringone induction, we calculated a fitting curve. Following the approach described in Nelson et al.(8), we used a dose response function and a logistic equation(1) for the data calculation of the receptor system. The luminescent response was proportional to the concentration of acetosyringone used for induction(3).
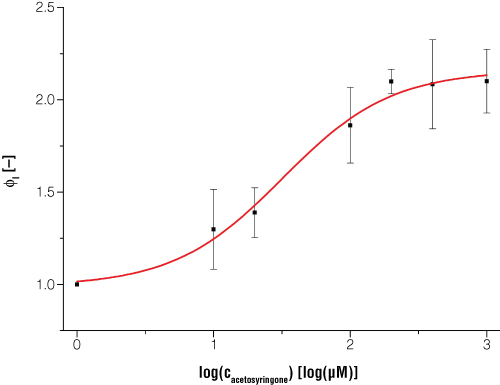
Figure 5. Expression of bioluminescence in E. coli is dependent on certain inducer concentrations when acetosyringone is the inducer. Dataset is normalized to a reference shaker flask cultivation without induction and to the time point t = 0. Every data point is the average value from three biological replicates. Switching point (50% of maximum induction) occurs at an acetosyringone concentration of 31.5µM, and at 100% induction the luciferase light signal is increased two times compared to the uninduced status.
Next we created three new BioBricks containing the virA, virG, or vir promoter. We also tried to create a light signal that is visible to the naked eye. For this purpose, we added genetic amplifiers (Cambridge, iGEM 2007, amplifier project) to our luciferase BioBrick. This amplified luciferase readout was coupled with the VirA/G signaling system to visualize the induction behavior. Although the amplifiers gave visible luciferase readout, the basal expression of the vir promoter was also amplified. Due to the strong amplification of both the basal expression and the induced expression, the VirA/G system could not be clearly distinguished from uninduced samples (Figure 6).

Discussion
In this project, we established an E. coli-based biosensor that is capable of sensing and measuring acetosyringone induction. Besides the functionality of the VirA/VirG two-component system in E. coli, which was shown previously by Jung et al.(9), we combined the signaling pathway with a luciferase-based readout system. We further demonstrated that the luciferase intensity correlated with the concentration of acetosyringone used to induce the cells. When this data was fitted to a dose-response curve, it was shown to be in close relation to the expected biological function of the receptor. The basal luciferase expression that was present in the uninduced system is the result of a leaking virB promoter. However, when the cells were induced with a saturating concentration of acetosyringone, the luciferase expression was up to twofold higher than for the uninduced state.
Furthermore, we provided eight new BioBrick parts to the database, which can be used in prospective projects.
Outlook
To design a novel biosensor using a directed evolution-driven approach to receptor modification, we established an error-prone PCR protocol as well as a screening system (data not shown). In the ongoing work, we are going to use an iterative process to optimize the receptor specificity and selectivity to closely related phenolic compounds (e.g., capsaicine).
Acknowledgements
The iGEM Project was supported by the Center for Biotechnology, CeBiTec at the University Bielefeld, Germany. Additional project funding was provided by: BIO.NRW Cluster Biotechnology of the federal state of North Rhine-Westphalia, Evonik GmbH, Promega GmbH, PlasmidFactory & Co. KG, and A.S.I.
References
- DeCleene, M. and DeLay, J. (1976) The host range of crown gall. Bot. Rev. 42, 389–466.
- Wolanin, P.M. et al. (2002) Histidine protein kinases: key signal transducers Genome Biol. 3, Reviews3013.
- Brencic, A. and Winans, S.C. (2005) Detection of and response to signals involved in host‐microbe Microbiol. Mol. Biol. Rev. 69, 155–194.
- Huang, T. et al. (1990) VirA, a corregulator of Ti‐specific virulence genes, is phosphorylated in vitro. J. Bacteriol. 172, 1142–1144.
- Jin, S.G. et al. (1990) Phosphorylation of the VirG protein of Agrobacterium tumefaciens by the autophosphorylated VirA protein: Essential role in biological activity of VirG. J. Bacteriol. 172, 4945–4950.
- Jin, S.G. et al. (1990) The regulatory VirG protein specifically binds to a cis‐acting regulatory sequence involved in transcriptional activation of Agrobacterium tumefaciens virulence genes. J. Bacteriol. 172, 531–537.
- Pazour, G.J. and Das, A. (1990) Characterization of the VirG binding site of Agrobacterium Nucleic Acids Res. 18, 6909.
- Pazour, G.J. and Das, A. (1990) VirG, an Agrobacterium tumefaciens transcriptional activator, initiates translation at a UUG codon and is a sequence‐specific DNA‐binding protein. J. Bacteriol. 172, 1241–1249.
- Jung, Y.C. et al. (2004) Mutants of Agrobacterium tumefaciens virG gene that activate transcription of vir promoter in Escherichia coli Curr. Microbiol. 49, 334.
Learn more about the Luciferase Assay System