Bioluminescent Nicotinamide Adenine Dinucleotide Detection Assays: Part II - Choosing the Right Product for Your Application
Promega Corporation
Publication Date: Revised May 2021
Abstract
The NAD(P)/NAD(P)H-Glo™ Assays are bioluminescent assays for the sensitive measurement of nicotinamide adenine dinucleotides. In this article we provide general guidelines for using these assays to measure enzyme activity in biochemical applications and for determining total or individual oxidized and reduced forms in cell-based applications.
Nicotinamide adenine dinucleotides (NAD, NADH, NADP, NADPH) are recognized as important key regulators of cellular metabolism with roles in maintenance of redox status, cell survival, cell signaling and inflammation under normal and pathological conditions. Growing evidence suggests that many human diseases are associated with abnormal metabolic states, and therefore targeting metabolism of these important cofactors might provide new therapeutic opportunities (1) (2). Understanding how these dinucleotides are regulated in cells and identifying assays that can accurately measure them has become increasingly important.
Maintaining the appropriate levels and balance between oxidized and reduced forms is important for cell survival and function. NAD is the only dinucleotide that is synthesized in cells de novo or through a salvage pathway. NADP is made from NAD by NAD kinase (NADK). NAD and NADH, as well as NADP and NADPH, are interchangeable through oxidation/reduction reactions catalyzed by NAD(P)H-dependent dehydrogenases. For example, in glycolysis, the tricarboxylic acid (TCA) cycle and fatty acid beta oxidation, NAD is used as a cofactor to produce NADH. While in mitochondria, NADH is used for ATP synthesis through the mitochondrial electron transport chain. Similarly, NADP is converted to NADPH in the pentose phosphate pathway, and NADPH is used by the cell for de novo fatty acid synthesis or regulation of redox status through the Nox, GSH and Trx pathways (3).
Unlike the metabolic redox reactions, which reversibly oxidize or reduce nicotinamide adenine dinucleotides, NAD biosynthetic and signaling pathways continuously produce and consume NAD. The levels of dinucleotides available in a cell continuously changes, resulting in the complex regulation and interaction of biosynthetic and signaling pathways that are tightly controlled by the levels and/or the ratio of the oxidized and reduced forms of nicotinamide adenine dinucleotides.
To better understand the roles of these important cofactors we need methods to: (1) Rapidly measure changes in the total amount of dinucleotides resulting from changes in their biosynthesis or consumption; (2) Measure the changes in individual dinucleotides and their ratio; and (3) Measure the activity of NAD(P)/NAD(P)H-producing and -consuming enzymes for new inhibitor/activator identification.
In a previous PubHub article (4), we introduced three bioluminescence assays developed for nicotinamide adenine dinucleotide detection: The NAD(P)H-Glo™ Detection System, the NAD/NADH-Glo™ and the NADP/NADPH-Glo™ Assays. In that article we described their underlying chemistry and features. Here we provide general guidance for applying these assays for measuring enzyme activity and for determining cellular levels of total or individual oxidized and reduced forms of dinucleotides.
Measuring Enzyme Activity and Screening for Inhibitors
All three assay systems can be used for measuring the activity of purified enzymes and inhibitor screening (Table 1). The simple protocols are compatible with automated and high-throughput workflows. The flexible assay chemistries are readily adaptable to 96-, 384- and 1,536-well plate formats and yield Z′ values >0.7 (data not shown). The use of the assays for measuring endogenous enzyme activity in cell or tissue lysates has not been tested.
The NAD(P)H-Glo™ Detection System (Cat.# G9061, G9062) can be used for in vitro biochemical characterization and inhibitor screening of dehydrogenases, a class of enzymes that are involved in reduction or oxidation of nicotinamide adenine dinucleotides. High sensitivity and low data variability permit measurement of low-activity enzymes or the setup of reactions at low NAD(P)H concentrations (e.g., for enzymes with low Km values for those dinucleotides). This assay can measure NAD(P)H production (increase in light signal) as well as NAD(P)H consumption (decrease in light signal).
Alternatively, to measure NAD(P)H consuming enzymes using an increase-in-signal approach, the NAD/NADH-Glo™ and NADP/NADPH-Glo™ Assays can be applied. Using this approach, unused NADH or NADPH substrate is degraded by acid followed by measuring the remaining NAD or NADP product. To extend the linear range for the increase-in-signal approach, we have developed protocols that extend the range up to 20µM NADH or NADPH. The increase-in-signal assay is more sensitive than the decreased-signal assay and detects 5% conversion of NAD(P)H to NAD(P) with a signal-to-background ratio >5 (data not shown).
Enzymes involved in NAD biosynthesis or NAD-dependent signaling pathways are important drug targets and can be screened using the NAD/NADH-Glo™ Assay. Since NADH is not present in the reactions, the NAD/NADH-Glo™ Luciferase Detection Reagent can be directly added to the sample to measure NAD production or consumption.
| Table 1. Application of the NAD(P)/NAD(P)H-Glo™ Assay Systems for Enzyme Activity Measurements. | ||||||
|---|---|---|---|---|---|---|
| Assay | Dehydrogenases and Oxidases | NAD-Dependent Enzymes | Assay Range | Comments | ||
| NAD(H)-Dependent | NADP(H)-Dependent | Biosynthetic | Signaling | |||
| NAD(P)H-Glo™ Detection System | yes | yes | no | no | 25nM–50µM | Use alkaline solution to stop enzyme activity.1,2 |
| NAD/NADH-Glo™ Assay | yes | no | yes | yes | 1–500nM | Use acid to stop enzyme activity and/or destroy reduced forms.3 Linearity can be extended up to 20µM.4 |
| NADP/NADPH-Glo™ Assay | no | yes | no | no | 1–500nM | Use acid to stop enzyme activity and/or destroy reduced forms.3 Linearity can be extended up to 20µM.4 |
|
1 Addition of NAD(P)H-Glo™ detection reagent might not stop enzyme activity. In this case, generation of NADH or NADPH will continue, and the luminescent signal will increase until all Reductase Substrate is consumed. 2 Use 50–200mM KOH or NaOH final concentration to stop enzyme activity (so that reduced dinucleotides remain stable). Neutralize, for example, using HCl/Trizma® solution before adding detection reagent. 3 Use 0.1N HCl final concentration to stop enzyme activity and destroy reduced forms. Neutralize, for example, using 0.5M Trizma® solution before adding detection reagent. 4 To extend linearity of NAD detection use tenfold less NAD Cycling Enzyme and half the concentration of NAD Cycling Substrate recommended in TM399. For NADP use fourfold less NADP Cycling Enzyme and eightfold less NADP Cycling Substrate than stated in TM400. |
||||||
Assessing Metabolic Health and Performing Rapid Inhibitor and Activator Screens in Cell Culture
Like ATP, NAD(P) and NAD(P)H are key metabolites and good indicators of cell metabolic health. Cell death results in rapid loss of metabolic activity and degradation of the dinucleotides. Therefore measurement of total NAD + NADH or NADP + NADPH can provide information regarding the metabolic health of a cell population. Additionally, measurement of total reduced and oxidized forms of dinucleotide pairs can be used for rapid inhibitor/activator screens, in particular for modulators of NAD biosynthesis or consumption by NAD-dependent enzymes (5).
For these applications, the measurement of total NAD + NADH or NADP + NADPH is necessary or preferred. However, most protocols involve sample preprocessing such that only individual dinucleotides are measured. We developed protocols that include the option of measuring total dinucleotide pairs, eliminating extra preprocessing steps for cases when measuring dinucleotides separately is unnecessary.
The NAD/NADH-Glo™ and NADP/NADPH-Glo™ Assays allow measurement of the total amount of each dinucleotide pair in a population of mammalian cells using a rapid “add-and-read” protocol. The assays are sensitive enough to detect dinucleotides in a well of cultured cells in a 96- or 384-well plate (Table 2). To minimize addition steps, the detection reagent was formulated to contain detergents for cell lysis and all of the enzymes necessary for the bioluminescent reaction.
These assays can be performed in multiplex with a fluorescent, nonmetabolic cytotoxicity assay to gain information about interactions between drug-induced changes in metabolic pathways and cell death (5).
| Table 2. Rapid Detection of Changes in Total Amount of Dinucleotides in Mammalian Cells. | |||
|---|---|---|---|
| Application | Assay to Use | Assay Range1 | Format |
| Measure Total NAD+NADH | NAD/NADH-Glo™ Assay | 500–25,000 cells/50µl per well | Add detection reagent to cells in complete medium2 at 1:1 ratio. |
| Measure Total NADP+NADPH | NADP/NADPH-Glo™ Assay | 500–12,000 cells/50µl per well | Add detection reagent to cells in complete medium2 at 1:1 ratio. |
|
1 At higher cell densities or in cells expressing high levels of NAD-consuming enzymes, the activity of endogenous proteins can reduce dinucleotide stability and interfere with detection. In those cases as well as for individual dinucleotide quantitation we recommend using the base/acid protocol presented in the next section. 2 Relative light output is influenced by media composition; therefore, for dinucleotide quantitation, the standard curves have to be performed in the same medium as the experiment. However, because dinucleotides are not stable in complete medium with serum, the solutions of standards should be prepared in PBS and added to the medium with serum after the addition of detection reagent. |
|||
Detecting Shifts in the Ratios of Oxidized and Reduced Forms of Dinucleotides to Study Dysregulation of Metabolic Pathways
Dysregulation of metabolic pathways often results in a shift of the cellular oxidative state due to changes in the ratio of oxidized and reduced forms of nicotinamide adenine dinucleotides (i.e., NAD/NADH and NADP/NADPH). This shift can be detected by determining these ratios, which requires measuring the oxidized and reduced forms separately.
Commonly used protocols for isolating oxidized and reduced forms often begin by lysing two separate samples of cells. One cell sample is lysed in base with heat (~65°C) to degrade the oxidized form, and the second cell sample is lysed in the presence of acid to degrade the reduced form. The base and acid samples are then each neutralized, usually with a strong acid and strong base, respectively, and measurements taken. Samples prepared using these methods can be compatible with the NAD(P)/NAD(P)H-Glo™ Assays. Specific acids and bases should first be tested and samples must be neutralized to ~pH 6.5–8.5 before assaying.
There are several challenges with these methods. First, since the cells are split into two samples before lysis, there is no guarantee that the cell samples are equivalent and that cell lysis is equally complete. Second, because there is little buffering in the system (neutralization involves adding acids to base and bases to acid), it is easy to overshoot the desired pH of the final solution in which measurements are made. Quite often the samples will not be sufficiently neutralized, resulting in unreliable measurements. Third, because most of these approaches require large amounts of starting material, the samples can be very viscous, especially after lysing in strong bases; therefore a centrifugation step is required before analyzing the supernatant.
For detection of individual dinucleotides using NAD/NADH-Glo™ and NADP/NADPH-Glo™ Assays, we developed an optimized protocol to address these challenges and take advantage of the greater sensitivity of the assays. The optimized protocol is summarized in Figure 1. Both oxidized and reduced forms are measured from a single sample that is split after lysis rather than before. Because the assays require fewer cells than other protocols, direct processing of cells in multiwell plates is possible. The need for tubes and centrifugation is eliminated, facilitating higher sample throughputs.
In this optimized protocol (Figure 1, Table 3), the starting cells can be in PBS or medium without serum. Media or other buffers with high buffering capacity are not recommended. The entire sample is lysed in bicarbonate buffer containing the detergent DTAB. This protocol can also be used with tissue samples (Table 3). Tissue samples require additional homogenization, preferably in PBS/bicarbonate/DTAB lysis buffer.
Because both oxidized and reduced forms are stable in bicarbonate buffer, it is possible to measure total NAD+NADH or NADP+NADPH at this stage using NAD/NADH-Glo™ and NADP/NADPH-Glo™ Assays, respectively. Also, if multiple samples have to be collected at different time points or stored before assaying, we recommend processing the samples up to this point and freezing them at –70°C.
For detection of individual dinucleotides, the sample is split after lysis and exposed to similar conditions to minimize variability. To measure the reduced form, one-half of the sample is heated in lysis buffer only; to measure the oxidized form, the other half of the sample is heated in the presence of acid. Next, both samples are neutralized and buffered with a premixed HCl/Trizma® or Trizma® solution, respectively. It is necessary to premix HCl with Trizma® because HCl added separately will result in immediate degradation of reduced forms. Additionally, if desired, the neutralization solution can be premixed with detection reagent to minimize addition steps.
Individual dinucleotides are measured by adding the appropriate detection reagents to the neutralized samples at a 1:1 ratio. The detection reagent can be added to the entire neutralized sample or an aliquot. For example, one aliquot can be taken for the NAD/NADH-Glo™ Assay and another for the NADP/NADPH-Glo™ Assay, so that all four dinucleotides can be measured using samples prepared from a single well of cells. Because both acid- and base-treated samples are in the same buffers after neutralization, the ratio can be calculated directly based on light signal (RLU) values. Dinucleotide concentrations can be calculated by including a standard curve in the experiment.
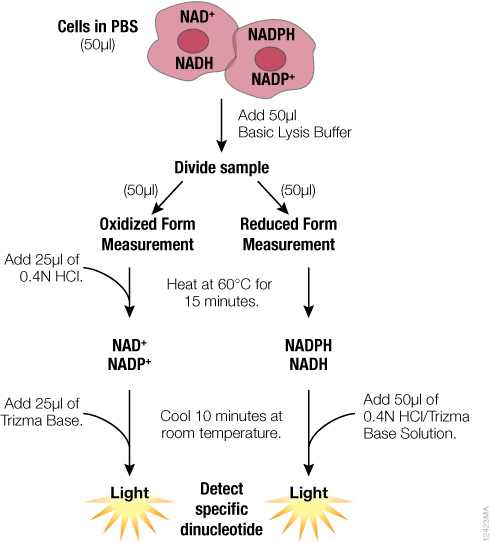
Figure 1. Schematic of the sample preparation protocol for measuring individual dinucleotides using the NAD(P)/NAD(P)H-Glo™ Assay Systems. Further description and formulation of the solutions can be found in technical manuals TM399 and TM400.
| Table 3. Recommended Assays for Detection of Individual Dinucleotides in Mammalian Cells and Tissues. | ||||
|---|---|---|---|---|
| Assay | Application | Samples | Assay Range | Comments |
| NAD/NADH-Glo™ Assay | Measuring total NAD+NADH; Measuring individual NAD and NADH; Determining NAD/NADH ratio | Mammalian Cells | Starting amount 2,000–100,000 cells/50µl PBS | After acid/base treatment dinucleotides will be in 100µl; take 20–100µl for detection.3,4 |
| NAD/NADH-Glo™ Assay | Measuring total NAD+NADH; Measuring individual NAD and NADH; Determining NAD/NADH ratio | Tissues | Starting amount 2–4mg protein /1ml homogenization buffer1 | Treat with acid/base in tubes or 96-well plate;2 take 20–100µl (10–100µg protein) for detection.3,4 |
| NADP/NADPH-Glo™ Assay | Measuring total NADP+NADPH; Measuring individual NADP and NADPH; Determining NADP/NADPH ratio | Mammalian Cells | Starting amount 2,000–100,000 cells/50µl PBS | After acid/base treatment dinucleotides will be in 100µl; take 20–100µl for detection.3,4 |
| NADP/NADPH-Glo™ Assay | Measuring total NADP+NADPH; Measuring individual NADP and NADPH; Determining NADP/NADPH ratio | Tissues | Starting amount 2–4mg protein/1ml homogenization buffer1 | Treat with acid/base in tubes or 96-well plate;2 take 20–100µl (10–100µg protein) for detection.3,4 |
|
1 We recommend homogenizing tissues in PBS/Bicarbonate/0.5% DTAB buffer using a manual tissue tearor or FastPrep® Systems (MP Biomedicals). The DTAB detergent in the extraction solution is compatible with Pierce 660nm Protein Assay with Ionic Detergent Compatibility Reagent (IDCR). 2 After homogenization, the samples can be taken directly for acid/base treatment; there is no need for centrifugation. The assay/base treatment can be performed in tubes or part of the sample can be transferred into 96-well plates for the treatment. 3 The recommendations are provided as general guidance. Since the level of dinucleotides will vary in different samples, different dilutions of samples have to be tested to make sure readouts are in the linear range of the detection system. 4 For quantitation, a standard curve can be generated using purified dinucleotide standards. The standards should be diluted in a solution identical to the final formulation of the neutralized samples (i.e., 1:1:1:1 ratio of PBS or medium: Bicarbonate Base + 1% DTAB: 0.4N HCl: 0.5M Trizma® base). |
||||
Summary
The NAD(P)/NAD(P)H-Glo™ family of assays provides robust, simple and flexible tools for cellular and biochemical analysis of nicotinamide adenine dinucleotides for a variety of experimental applications. These assays can be used to measure the activity of purified enzymes, perform rapid screens for inhibitors or activators of NAD-dependent enzymes, assess the metabolic health of cells, and study the dysregulation of metabolic pathways.
References
- DeBerardinis, R.J. and Thompson, C.B. (2012) Cellular metabolism and disease: What do metabolic outliers teach us? Cell 148, 1132–44.
- Mouchiroud, L., Houtkooper, R.H. and Auwerx, J. (2013) NAD+ metabolism: A therapeutic target for age-related metabolic disease. Crit. Rev. Biochem. Mol. Biol. 48, 397–408.
- Nakamura, M., Bhatnagar, A. and Sadoshima, J. (2012) Overview of pyridine nucleotides review series. Circulation Research 111, 604–10.
- Leippe, D., Sobol, M., Hooper, K., Arduengo, M. and Vidugiriene, J. (2014) Bioluminescent Nicotinamide Adenine Dinucleotide Assays Part I: Technology and Features Promega PubHub
- Vidugiriene, J. et al. (in preparation)
Learn more about the NAD(P)H-Glo™ Detection System