Exploring RAS and RAF Target Engagement: Live-Cell Insights into Molecular Specificity
Introduction
RAS proteins, pivotal as biomolecular switches, are essential in regulating intracellular activities such as cell proliferation and survival. Their frequent mutations in human cancers underscore their importance in both basic research and drug discovery. Historically, targeting RAS proteins with small molecule drugs has been challenging due to the absence of conventional small molecule binding pockets. Nevertheless, significant progress has been made, notably with the accelerated FDA approval of a covalent inhibitor (AMG 510 or Lumakras) targeting the KRAS-G12C mutant for specific lung cancer treatment. This breakthrough, along with the development of other allele-specific inhibitors (1) as well as the application of newer modalities aimed at targeting RAS proteins for degradation (2), illustrates the potential of directly targeting mutant RAS proteins, either alone or in combination.
Given the historic difficulties in directly targeting RAS, research efforts have also included downstream effectors like RAF kinases, which play a critical role in the RAS-RAF-MEK-ERK signaling pathway. RAF kinases, particularly BRAF variants, have been the focus of drug discovery due to their oncogenic role in various cancers. However, first-generation BRAF-selective kinase inhibitors, while effective in BRAF mutant cancer cells, result in ERK activation in BRAF wild type and RAS mutant cells, leading to the "RAF inhibitor paradox” (3,4) and illustrating further the challenges in tackling this pathway.
Both RAS and RAF protein families consist of closely related isoforms with additional diversity stemming from clinically significant hotspot mutations, especially prevalent within RAS. Compounding this complexity, the signaling functions of both RAS and RAF are contingent upon their ability to form cellular protein complexes, essential for triggering downstream signaling cascades. Given the intricacy of these pivotal nodes within the RAS-RAF-MEK-ERK pathway, there is need for advanced model systems that enable the thorough assessment of drug molecules in an environment that closely replicates the natural biological context in which these proteins operate.
Although various cellular approaches, such as measuring ERK phosphorylation, can gauge the effects of compounds targeting RAS or RAF on downstream MAPK signaling, a comprehensive understanding of direct drug-target interactions is crucial for advancing small molecule development programs. While cell-free/biochemical methods often provide insights into small molecule binding and specificity, these findings may not always align with cellular biomarkers of MAPK activity, posing challenges for decision-making processes. This article discusses the development of novel target engagement methodologies that allow for the detailed analysis of compound binding affinity and specificity to RAS and RAF protein complexes in live cells, creating new tools for researchers studying this clinically significant signaling pathway.
Adapting NanoBRET® Technology to Measure Compound Engagement with RAS and RAF Protein Complexes
NanoBRET® Target Engagement (TE) technology is a live-cell solution designed to measure binding between a target protein and small molecule using an energy transfer technique known as bioluminescence resonance energy transfer (BRET). Typically, this energy transfer is based on two elements: cellular expression of the target protein fused to NanoLuc® Luciferase and a cell-permeable fluorescent NanoBRET® tracer, which binds reversibly to the target protein. Displacement of the labeled tracer allows for a specific and quantitative measure of compound binding affinity.
Given the intrinsic complexity of RAS and RAF proteins, which function intracellularly in a dimerized state, the NanoBRET® TE system has been modified to enable the determination of target engagement with a protein-protein complex (Figure 1). This adaptation involves the use of NanoLuc® Binary Technology (NanoBiT®), a structural complementation reporter system that consists of a Large BiT (LgBiT; 18kDa) subunit and a small complimentary peptide, to create the energy donor used in the BRET reaction. In the NanoBRET® TE Intracellular RAS and RAF assays, the small complementary peptide is called SmBiT(q), which has low affinity for the LgBiT subunit. LgBiT and SmBiT(q) are fused to the full-length RAS or RAF proteins being studied and co-expressed in cells. When the target proteins interact and form multimers, the LgBiT and SmBiT(q) subunits come together to form an active luciferase enzyme and, in the presence of a luciferase substrate, generate a bright luminescent signal that serves as the BRET energy donor. This setup enables the direct measurement of compound binding to the cellular protein complex.
Leveraging NanoBRET® TE Assays for Specificity Analysis in Compound Binding to RAS and RAF Variants
When targeting RAS and RAF proteins with small molecule inhibitors or degraders, it can be valuable to not only understand the potency of test compound binding against a specific variant being targeted, but also the potential engagement of the test compound across other variants or known mutations. Understanding the compound selectivity profile can aid in interpretation of downstream signaling results, facilitate further tuning of compound selectivity, and help to better predict how the compound will perform in clinical settings.
The NanoBRET® TE Intracellular RAS Assay enables the assessment of compound selectivity across a suite of 15 different RAS variants, including KRAS, HRAS and hotspot mutants. This includes mutations such as the KRAS(Y96D) variant, to evaluate the susceptibility of test compounds to commonly observed resistance mutations. This approach has recently been used to provide valuable insights into the potential clinical utility of inhibitors targeting the switch II pocket (5). The assay set was designed using a single tracer at a consistent concentration for all variants, creating a simplified workflow for profiling test compound selectivity and engagement potency across a range of RAS proteins.
Figure 2 illustrates the selectivity profiling of four distinct compounds against a suite of KRAS and HRAS hotspot mutants. By co-transfecting the LgBiT and SmBiT(q) fusion vectors for each target protein and conducting dose-response analyses, IC50 potency values for each compound across RAS variants are ascertainable. This data reveals the differential engagement potency of compounds like SI/II-pocket ligand BI-2852 and SII-pocket ligands Sotorasib, Adagrasib, and MRTX1133 across various RAS forms, highlighting their respective potencies and selectivities. BI-2852 demonstrates competition of BRET signal for all RAS variants with similar potency, making it an ideal positive control for target engagement across the RAS variants.

A key feature of this alternative NanoBiT® donor is its sensitivity to the Extracellular NanoLuc® Inhibitor, which is enabled by the use of the SmBiT(q) peptide. This ensures that the luminescent signal used as the energy donor originates only from live cells, increasing specificity by reducing background noise from compromised cells or cellular debris. The assays quantify target engagement and compound affinity by observing the competition between a fluorescent NanoBRET® tracer and test compounds for binding to RAS or RAF complexes, with a decrease in the BRET signal indicating successful target engagement. Importantly, this iteration of NanoBRET® TE creates a conditional BRET donor that will only form when the target proteins form a complex, providing a method to quantify apparent intracellular affinity of a test compound for full-length multimeric RAS and RAF proteins inside living cells.

Similarly, the NanoBRET® TE Intracellular RAF Dimer Assays facilitates the evaluation of compound selectivity across different RAF dimer configurations, enhancing the understanding of compound engagement and selectivity within RAF complexes . Notably, the RAF dimer assays can be performed in the presence of constitutively active (untagged) KRAS(G12C) mutant isoform to replicate high KRAS activity conditions and promote complex formation (Figure 3). Compound target engagement can be studied on BRAF and CRAF homodimers or the assay can be used to evaluate compound binding to a specific RAF protomer within a heterodimeric complex. In this assay configuration, the primary RAF protomer of interest is paired with a second mutant RAF protein [BRAF(A481F) or CRAF(A373F)]. These alanine-to-phenylalanine mutations are pseudokinase mutations that prevent tracer binding to that specific RAF protomer but maintain the ability to form the RAF heterodimeric complex. This approach enables the assessment of compound binding specifically to the ARAF, BRAF, and CRAF protomers when they are present within heterodimeric complexes.
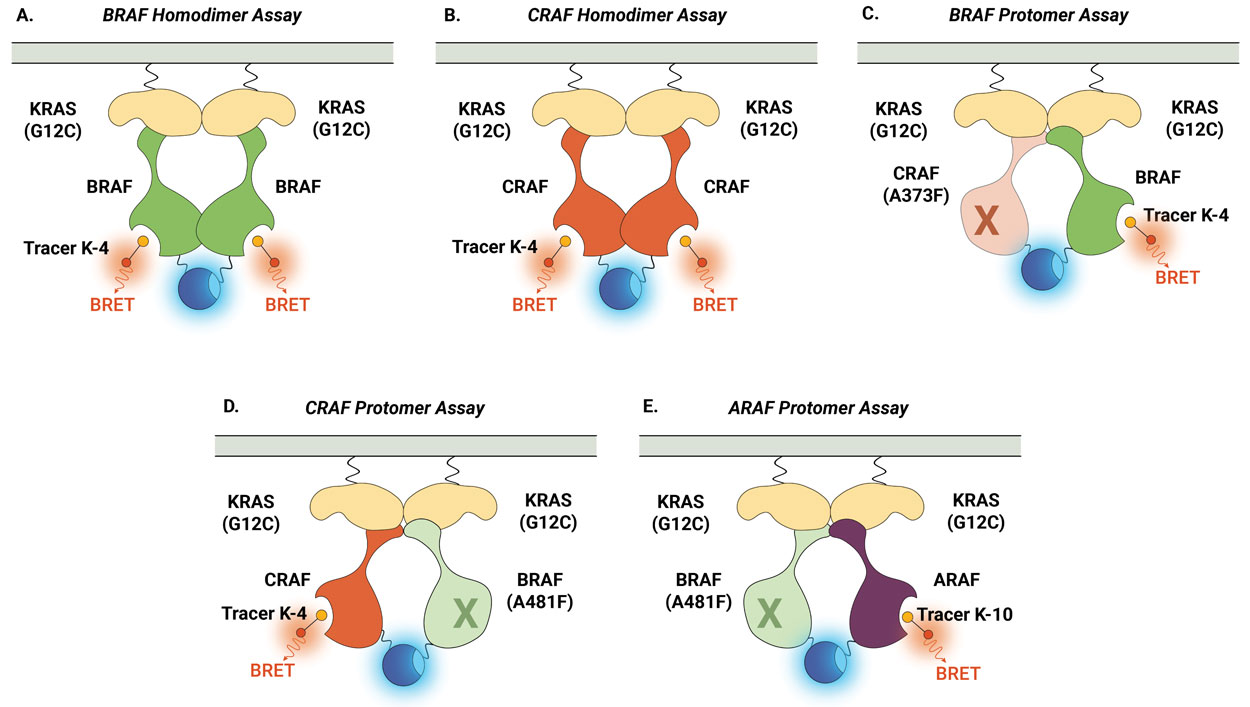
Figure 4 demonstrates how the NanoBRET® TE Intracellular RAF Dimer Assays can be used to profile compound activity across multiple RAF configurations and understand protomer selectivity. Shown here are examples of activity profiles for RAF dimer inhibitor TAK-632 (Panel A) and LXH254 (Panel B). Both inhibitors can engage all RAF dimers, but both compounds show weaker affinity for CRAF within homodimers and for the ARAF protomer. Recently, this approach was used to characterize the protomer activity of a collection of type II RAF inhibitors and demonstrated that all tested inhibitors displayed reduced affinity for the ARAF protomer. However, only ARAF protomer engagement, but not CRAF or BRAF engagement, corresponded with the inhibition of RAS-RAF-mediated signaling (6).

The RAS-RAF-MEK-ERK signaling pathway remains both a beacon of promise and a challenge in the realm of drug discovery. The introduction of advanced assay technologies, like the live-cell NanoBRET® TE Intracellular Assays for RAS and RAF, provide a step forward in the creation of more biologically relevant models that can be added to the researcher toolbox. Such tools will be instrumental in deepening our understanding of the complex biology underpinning cancer progression and therapeutic resistance, helping to pave the way for the innovation of novel drugs aimed at these once undruggable targets.
References
- Moore, A. R. et al. (2020). RAS-targeted therapies: is the undruggable drugged? Nat. Rev. Drug Discov. 19(8), 533–552.
- Bery, N. et al. (2020). A potent KRAS macromolecule degrader specifically targeting tumours with mutant KRAS. Nat. Comm. 11, 3233.
- Cook, F.A. and Cook, S.J. (2020). Inhibition of RAF dimers: it takes two to tango. Biochem. Soc. Trans. 49, 237–251.
- Cox, A.D. and Der, C.J. (2010). The Raf inhibitor paradox: unexpected consequences of targeted drugs. Cancer Cell. 17, 221–223.
- Vasta, J.D. et al. (2022). KRAS is vulnerable to reversible switch-II pocket engagement in cells. Nat. Chem. Biol. 18, 596–604.
- Vasta, J.D. et al. (2023). Protomer selectivity of type II RAF inhibitors within the RAS/RAF complex. Cell Chem. Bio. 30(11), 1354–1365.
Learn more about our NanoBRET® TE Intracellular RAS and RAF Dimer Assays
Related Resources
RAS Drug Discovery
Learn more about assays and technologies that facilitate RAS drug discovery.
Publication: Lumit p-ERK
View this publication: "Profiling oncogenic KRAS mutant drugs with a cell-based Lumit p-ERK immunoassay"