High Protein Yield and Purity with the HaloTag® Protein Purification System
Kate Qin Zhao, Jackie Kinney, Rachel Friedman Ohana and Marjeta Urh
Promega Corporation
Publication Date: 2010
Abstract
Introduction
HaloTag® Technology is based on a 34kDa monomeric protein tag modified from a bacterial dehalogenase, designed to covalently bind to synthetic HaloTag® ligands (1,2). A variety of HaloTag® ligands are available for in vivo and in vitro applications, such as fluorescent ligands for protein detection and cell imaging, and HaloLink™ Resin for isolating protein complexes as well as purification.
HaloTag® Technology has been optimized for compatibility with many protein expression systems including E. coli, mammalian cells and cell‑free expression systems. When studied with more than 20 difficult‑to‑express human proteins, the HaloTag® fusion tag has been shown to outperform GST, MBP and His•Tag® in producing soluble fusion proteins and purifying target proteins of higher yield and purity from E. coli (3).
When studied with more than 20 difficult‑to‑express human proteins, the HaloTag® fusion tag has been shown to outperform GST, MBP, and His•Tag in producing soluble fusion proteins and purifying target proteins of higher yield and purity from E. coli.
The HaloTag® Protein Purification System has three components: HaloLink™ Resin, TEV Protease and HisLink™ Resin. A streamlined protocol has been developed around these components to complete the following four steps in purification (see also Figure 1):
- Covalent binding for efficient and specific capture of a HaloTag® fusion protein onto HaloLink™ Resin. Covalent capture makes it possible to recover proteins expressed even at low levels.
- Wash away unbound protein contaminants; stringent washes are possible with minimal loss after the HaloTag® fusion protein is covalently attached to HaloLink™ Resin.
- Release the target protein from the HaloTag® protein tag by on-resin TEV Protease cleavage, leaving the HaloTag® protein tag itself permanently attached to the HaloLink™ Resin.
- The TEV Protease included in the system [with an N-terminal (HQ)3 tag] is removed from target of interest by HisLink™ Resin.
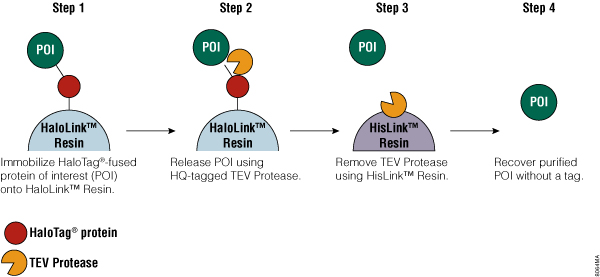
The result is purified protein of interest, typically recovered with high yield and purity and without HaloTag® protein tag carryover. One simple buffer, the HaloTag® purification buffer (HT buffer: 50mM HEPES (pH 7.5), 150mM NaCl with optional additives 1mM DTT, 0.5mM EDTA, and 0.005% IGEPAL® CA-630; HT buffer referred to in this article, contained all the optional additives), is compatible with all purification steps. If needed, the system can also be optimized to include stringent washing steps by altering buffer compositions to remove recalcitrant contaminants (see HaloTag® Protein Purification System Technical Manual #TM312 for details).
Using HaloTag® Technology in E. coli‑Based Expression and Purification
HaloTag® protein purification can be applied to proteins expressed in multiple systems. The HaloTag Protein Purification System Technical Manual #TM312, covers conditions developed and optimized for HaloTag® fusions expressed in E. coli, with additional recommendations for mammalian cells. Table 1 contains a quick guide to basic steps and required materials for E. coli-based HaloTag® expression and purification.
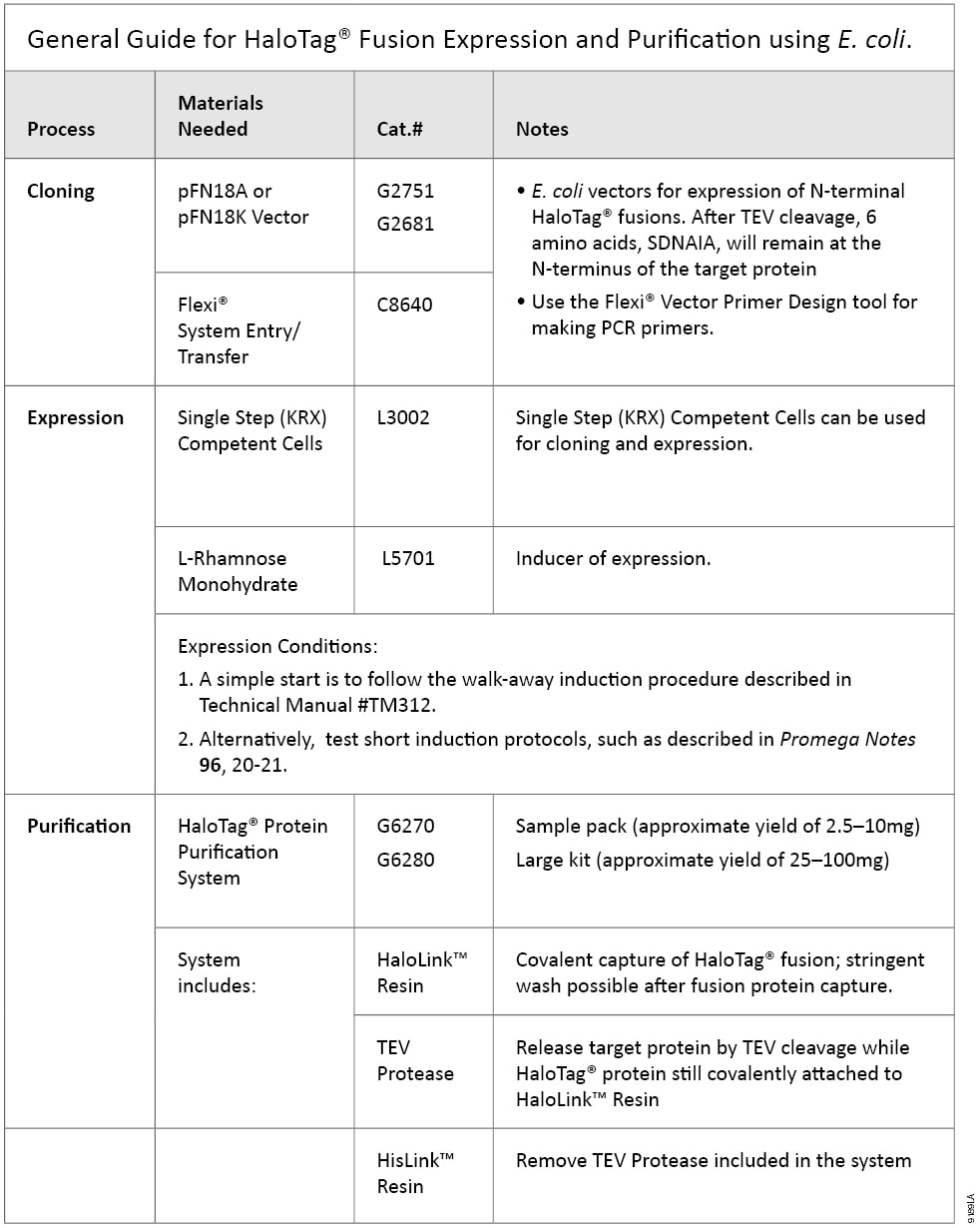
Features of HaloTag® Protein Purification System
In equilibrium-based affinity purification, protein is constantly exchanged between two states, either bound or not bound to the affinity resin (e.g., GST to GSH resin and His•Tag® to Ni2+-charged solid supports). This can lead to issues such as reduced binding efficiency when the fusion protein expression is low and the loss of protein targets during extensive washing. Additionally, large fusion tags, such as GST and MBP, often need to be removed from the protein of interest, usually by specific protease cleavage followed by affinity removal of the free tags. This latter step can be problematic, as the tags themselves are hard to eliminate and thus become a contaminant in the final purified protein.
HaloTag® technology-based purification, because of its covalent protein capture, provides solutions to these common problems (3):
- Covalent protein capture renders the immobilization process irreversible, so that the binding of fusion proteins is less affected by expression level.
- Extensive and stringent washes are possible with minimal loss of target protein, because;
- HaloTag® itself will not elute with target of interest, as it is permanently bound and remains on the HaloLink™ Resin after on-column TEV Protease cleavage. Upon TEV Protease removal, the target protein, typically of high yield and purity, is recovered.
Methods
The benefits of HaloTag® technology-based expression and purification in E. coli are demonstrated in the following three examples. All purifications were performed using a batch/column combination method as described in TM312. Specifically, cells from 50ml of expression culture were collected, resuspended in 5ml of HT buffer, disrupted by sonication and centrifuged at 10,000 × g, 4°C for 30 minutes to collect soluble supernatant; a ratio of 5ml of expression cell lysate supernatant to 1ml of settled HaloLink™ Resin was used. The amount of TEV Protease used was about 300units/ml of settled HaloLink™ Resin, and 50µl of 50% HisLink™ Resin was used to remove TEV Protease. The ratios are listed in Table 2.
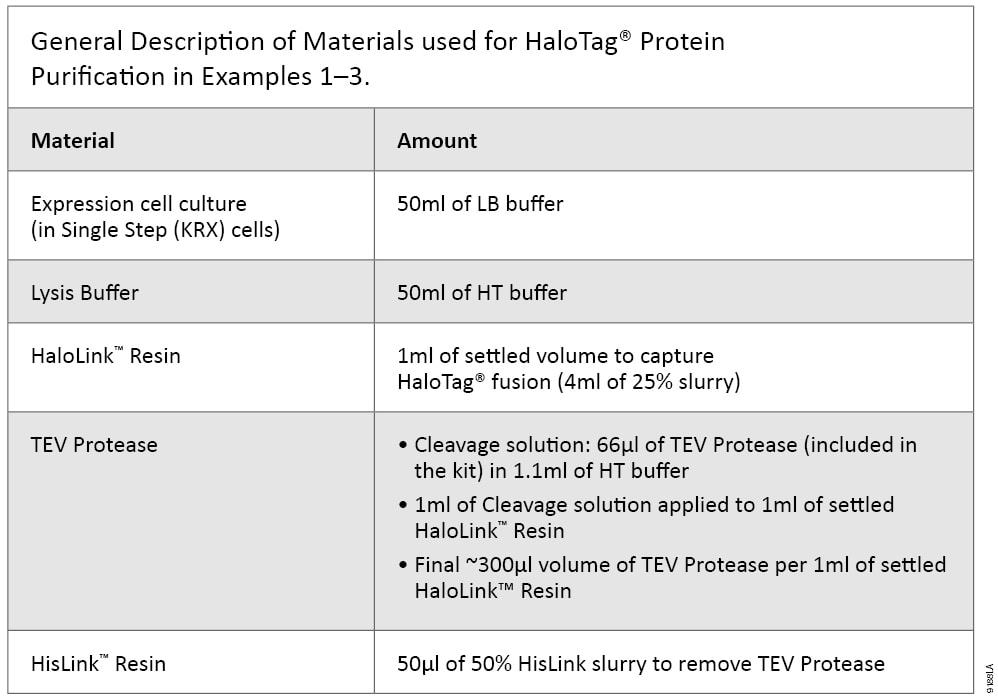
We compared samples from different purification steps using SDS-PAGE and/or an activity assay when applicable. The samples analyzed are described below with indicated nomenclature:
- S: Soluble cell lysate supernatant after cell lysis and high-speed centrifugation; this sample contains soluble fusion protein.
- S-Tev: Soluble cell lysate supernatant treated by TEV Protease; this sample has HaloTag® protein as well as untagged target protein, which is the reference for total recoverable target.
- FT: Flowthrough; this sample has unbound fusion protein after capture by HaloLink™ Resin, indicating binding efficiency when compared to the fusion protein in sample S.
- E1: Recovered target protein after on-resin Tev cleavage; this sample has untagged target as well as TEV Protease.
- E2: Final purified protein after removing TEV Protease.
The proteins are concentrated ~2.5-fold in E1 and E2 as compared to other fractions, since 2ml of the elutions is collected compared to 5ml of the starting cell lysate. This concentrating factor is applied in normalizing data analysis.
Results
Example 1: High Protein Recovery
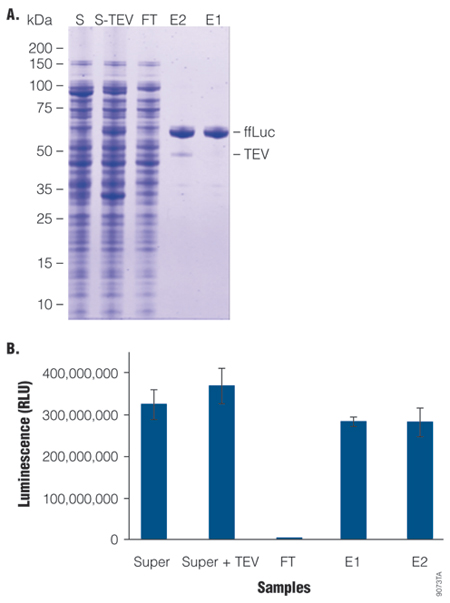
This example demonstrates high protein recovery, by analyzing the final protein yield of firefly luciferase (ffLuc) compared to that in the starting crude cell lysate. Protein recovery was analyzed by comparing the band intensities of ffLuc in E2 to that in S-Tev on a Coomassie®-stained SDS-PAGE analysis as well as by assaying ffLuc activity in the samples.
Firefly luciferase (ffLuc) was purified from 50ml of culture using 1ml of settled HaloLink™ Resin by batch/column combination method as described in TM312 and in Table 2. Purified protein was of high purity (>90%) as judged by SDS-PAGE analysis, and of high recovery as determined by comparing the Coomassie®-stained ffLuc band intensity in E2 to that in S-Tev. Active protein recovery was greater than 75% of the starting material (E2 compared to S-TEV), suggesting efficient capture of fusion protein and protein release by TEV Protease cleavage.
Example 2: Recovery of Proteins From Low Expression-Mimicking, Diluted Lysates
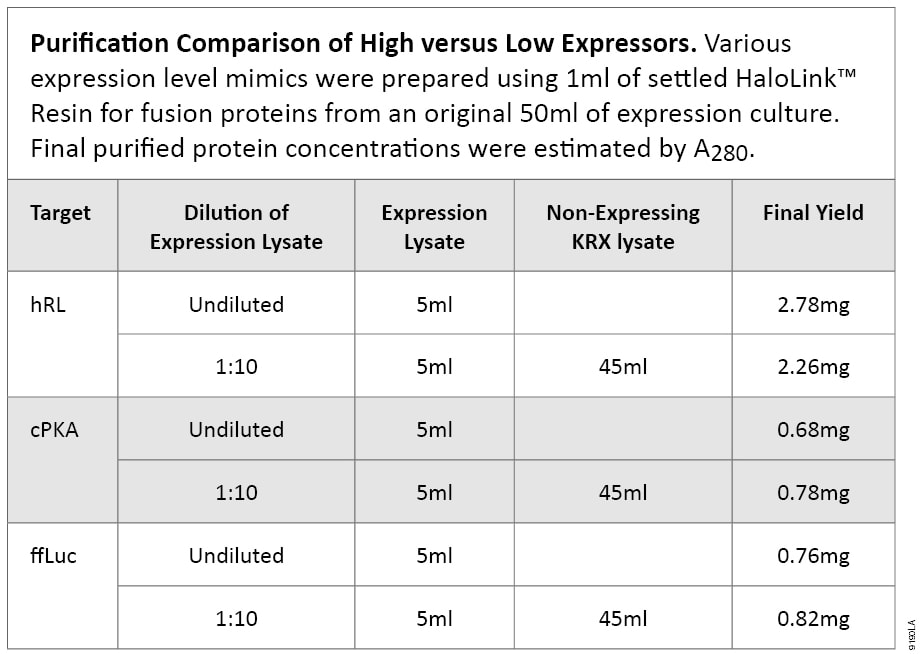
To demonstrate the ability of HaloTag® technology to purify proteins of low abundance, we compared purification from a tenfold diluted lysate to that from undiluted expression cell lysate. For the dilution analysis, we used nonexpressing cell lysate, prepared from Single Step (KRX) Competent Cells without a construct, where 200ml of KRX cell lysate was made from 2L of culture. The diluted sample represents a low-expressing fusion of 10% of the original expression lysate.
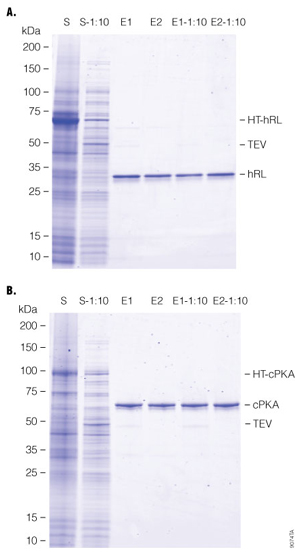
All purifications essentially are equivalent in terms of total amount of fusion protein applied to 1ml of settled HaloLink™ Resin except for the concentration of fusion protein in the lysate. Three targets were tested: ffLuc, hRL (humanized Renilla luciferase) and cPKA (human catalytic subunit of protein kinase A). In all cases, similar protein yields were obtained, when the same amount of fusion protein was applied to a set amount of HaloLink™ Resin. Table 3 shows starting materials and final protein yields. A representative purification comparison by SDS-PAGE is shown in Figure 3.
Example 3: Removal of E. coli Chaperonins: An Example of GroEL
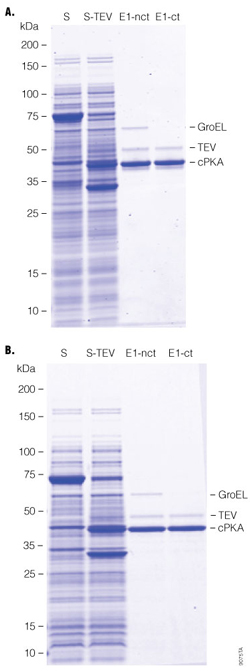
E. coli chaperonins (e.g., GroEL and DnaK) can assist protein folding (4). They are often copurified with recombinant proteins from E. coli and need to be removed by affinity chromatography. In this example, we successfully removed GroEL from human cPKA by two methods:
- The lysate was pretreated with a mixture of ATP/Mg2+ to dissociated chaperonin/protein complex prior to binding to HaloLink™ Resin, where HaloTag® protein capture was not affected by the treatment; and
- Chaperonin was removed on-column with ATP/Mg2+ after fusion protein binding to HaloLink™ Resin, where covalent capture ensured minimal protein loss during this treatment. The final proteins recovered were similar in both cases (Figure 4), demonstrating the versatility of the HaloTag® Protein Purification System.
Summary
HaloTag®-mediated protein purification is based on covalent immobilization, which enables efficient immobilization even for low abundance fusion proteins. This covalent immobilization also makes extensive washing possible for removing contaminants. HaloTag® technology has been shown to enhance soluble protein expression in E. coli (3). Thus the HaloTag®-based expression and purification strategy is a convenient and reliable method for the recovery of high-yield, pure, fusion tag-free protein of interest.
References
- Los, G.V. and Wood, K. (2007) The HaloTag: A novel technology for cell imaging and protein analysis. Methods Mol. Biol. 356, 195–208.
- Los, G.V. et al. (2008) HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 3, 373–82.
- Ohana, R.F. et al. (2009) HaloTag7: A genetically engineered tag that enhances bacterial expression of soluble proteins and improves protein purification. Protein Expr. Purif. 68, 110–20.
- Horwich, A.L. and Fenton, W.A. (2009) Chaperonin-mediated protein folding: Using a central cavity to kinetically assist polypeptide chain folding. Q. Rev. Biophys. 42, 83–116.
Learn more about the HaloTag® Protein Purification System