Multiplexing Cell-Based Assays: Get More Biologically Relevant Data
Kyle Hooper
Promega Corporation
Publication Date: 2011
Abstract
This article is an overview of the Promega cell-based assays that can be combined to measure multiple aspects of a response to treatment in cultured cells or normalize the experimental data to a control. Assessing multiple cellular parameters of a single sample well (e.g., apoptosis and cytotoxicity) provides more complete information about the event being measured and minimizes variability.
Introduction
The term “multiplexing” simply means gathering more than one set of data from the same sample. The key benefit of multiplexing is that you gain a better understanding of the event you are measuring in the context of another parameter, minimizing faulty interpretation of data or ambiguity from data sets. Multiplexing insures that the variables for a sample are the same for all the assays and that new variables are not introduced through replicate plating. Multiplexing gives you a way to normalize your data to a particular parameter, for instance coupling like the ratio of an activity to the number of cells in the well.
To multiplex two or more assays, the assays must meet certain criteria: the signals for the multiple assays must be spectrally or temporally distinct, the assay chemistries must be compatible, and the assays must fit into the same well or be easily separated. For example, cell-based assays can be performed in multiplex by combining both fluorescent and bioluminescent assays. The assays are read sequentially (Figure 1). Reading the different assays is easiest if you have a multimode plate reader such as the GloMax® Multi+ Instrument (1) , but can be accomplished by transferring the assay plate between dedicated fluorometers and luminometers.
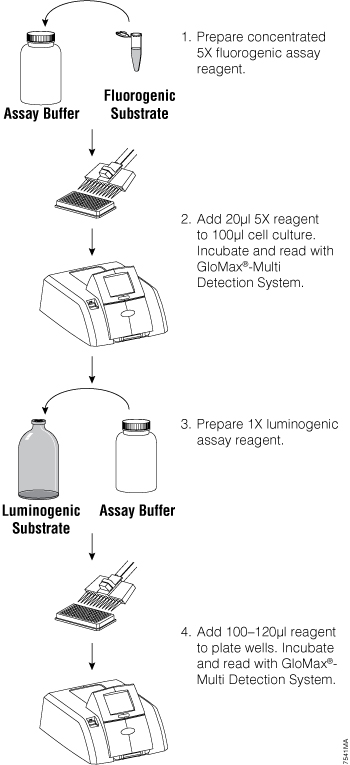
Figure 1. General fluorescent/luminescent multiplex protocol overview. From Zakowicz, H. et al. (2008) Measuring Cell Health and Viability Sequentially by Same-Well Multiplexing Using the GloMax®-Multi Detection System. Promega Notes 99, 25–8.
Multiplexing is not a new idea. When researchers first began using reporter assays, it became clear that a treatment can affect the general transcriptional activity of a cell without stimulating a specific promoter. Likewise, a treatment could kill the cell, and as a result promoter activity would appear to decrease. Researchers sought to use a control reporter that would not change with the treatment but remain constant in the experiment. They would make a cell lysate, split it and measure the experimental reporter (usually firefly luciferase) with one portion of the lysate and the control reporter (usually β-galactosidase) with the other. The ratio of the activity of the experimental to the control reporter would indicate whether a treatment was specific to a promoter or affected the whole cell. Splitting extracts was not ideal because variability could be introduced from assaying two separate samples. To solve this problem, the Dual-Luciferase® Reporter Assay System was developed, allowing two different luciferases to be measured in the same sample, one after the other. A sample of the extract is first assayed for firefly luciferase with the Luciferase Assay Reagent II. Stop & Glo® Reagent is then added to quench firefly luciferase and provide the substrate for the control reporter, Renilla luciferase. The multiplexing assay is made possible because the two luciferases have distinct assay chemistries, and the two assays can be measured separately using the same sample in the same well (2) .
Many researchers continue to use single-reporter assays for various reasons. For example, when using a stably transfected luciferase reporter cell line, there may be no need to control for transfection efficiency, and a single reporter may be sufficient. However, Dual-Luciferase® reporter assays also control for changes in cell viability during the course of the treatment (3) . When using a single-reporter assay, biological relevance is enhanced when you demonstrate concordance between a change in cell viability (fluorescent) and a change in genetic reporter (luminescent) activity as the direct result of cell cytotoxicity. If only a single reporter activity is measured, the effect of experimental treatment on cell health may be overlooked. The CellTiter-Fluor™ Cell Viability Assay (4) can be easily multiplexed upstream of single-reporter gene assays like the ONE-Glo™ Luciferase Assay System to account for changes in viability and normalize the data (Figure 2). The CellTiter-Fluor™ Assay is a fluorescent cell viability assay ideally suited for multiplexing with bioluminescent assays because it exhibits no intrinsic color quenching, which can limit luminescent assay sensitivity.
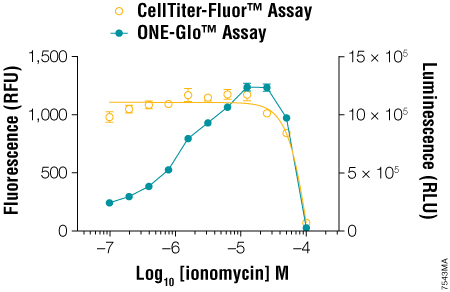
Figure 2. Multiplexing the CellTiter-Fluor™ Cell Viability and ONE-Glo™ Luciferase Assays. GloResponse™ NFAT-RE-luc2P HEK293 cells (10,000 cells/well, 100μl/well) were treated with serial dilutions of ionomycin in DMEM/10% FBS (containing hygromycin B and PMA, which is a known inducer of NFAT transcriptional activity). After 6 hours at 37°C, CellTiter-Fluor™ Reagent was prepared at 5X concentration and added to the wells in 20μl volumes. After a 30-minute incubation at 37°C, the fluorescence was measured on the GloMax®-Multi Detection System using the AFC (405nmEx/495–505nmEm) optical kit. ONE-Glo™ Reagent (100μl/well) was prepared and added to the wells, followed by luminescence measurement using the instrument’s luminescence module. From Zakowicz, H. et al. (2008) Measuring Cell Health and Viability Sequentially by Same-Well Multiplexing Using the GloMax®-Multi Detection System. Promega Notes 99, 25–8.
Two independent assays confirming the same event can provide more confidence that the event you are observing is real. The MultiTox-Fluor and MultiTox-Glo Multiplex Cytotoxicity assays were designed to measure both live cells and dead cells in a single well (1) (2) . For a cytotoxic event to occur, the viable cell reading must decrease while the dead cell reading must increase. Changes in one parameter without the cocommittant change in the other parameter suggest that something else was happening in the cells; both must occur. In Figure 3, true cytotoxic events are apparent due to coincident increases in dead-cell signal and decreases in live-cell signal. Events like fluorescent interference by the test compound and color quenching are easily identified as false cytotoxic events since there is no cocommittant change in the partner signal (Figure 3).
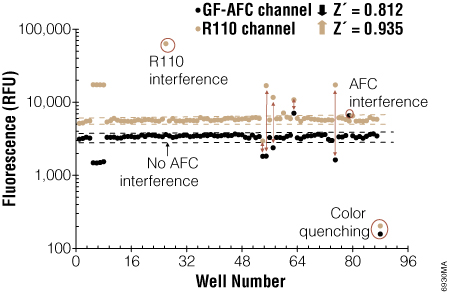
Figure 3. Confirming cytotoxicity by measuring changes in viable cells and dead cells with the MultiTox-Fluor Multiplex Cytotoxicity Assay. At the far right are control untreated cells and the next set are control 100% dead cells showing the maximum signal from the dead cell assay (R110 Channel) and minimal signal from the live cell assay (GF-AFC channel). True cytotoxic events are marked by an increase in the dead cell signal and a decrease in the live cell signal. From Niles, A. et al. (2007) Using Protease Biomarkers to Measure Viability and Cytotoxicity. Cell Notes 19, 16–20.
The MultiTox-Fluor Multiplex Cytotoxicity Assay is also useful for multiplexing with bioluminescent assays because it provides in a single addition two fluorescent assays with spectrally distinct signals. You can easily add the MultiTox-Fluor Assay upstream of a bioluminescent assay to gain insight into the state of cellular health. Directions for modifying the standard MultiTox-Fluor Assay for multiplexing are included in Technical Bulletin #TB348. The example in the Technical Bulletin multiplexes the MultiTox-Fluor Assay upstream of the Caspase-Glo® 3/7 Assay. This combination pushes the concept of positively identifying a cytotoxic event with concurrent monitoring of caspase activation to identify the mechanism of cytotoxicity, in this case caspase activation associated with apoptosis (Figure 4). An example of how a researcher used this combination can be found in Apolloni, S. et al. (5) .
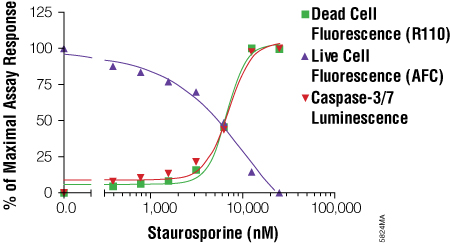
Figure 4. Using the MultiTox-Fluor Multiplex Cytotoxicity Assay with the Caspase-Glo® 3/7 Assay for a triplex assessment in an apoptosis model system. LN-18 cells were plated at a density of 10,000 cells per well in 50μl volumes of MEM + 10% fetal bovine serum and allowed to attach overnight. Staurosporine was twofold serially diluted and added to wells in 50μl volumes. The plate was incubated at 37°C in 5% CO2 for 6 hours. MultiTox-Fluor Reagent was prepared as 10μl Substrate in 1ml Assay Buffer, and 10μl was used. The plate was mixed and incubated for 30 minutes at 37°C. Fluorescence was read on a BMG PolarStar plate reader. Caspase-Glo® 3/7 Reagent was then added in an additional 100μl volume, and luminescence measured after a 10-minute incubation. The resulting signals were normalized to a percentage of maximal response and plotted using GraphPad Prism® software. From the MultiTox-Fluor Multiplex Cytotoxicity Assay Technical Bulletin #TB348.
Measuring viability, cytotoxicity and caspase activation is a powerful tool to determine the mechanism of cell death. The ApoTox-Glo™ Triplex Assay encompasses all three parameters in a unified protocol. The system monitors caspase activation in the context of changes in membrane integrity. Using this assay, you can differentiate necrosis from various stages of apoptotic cell death (6) . Several researchers have already incorporated this system into their research (7) (8) (9) . Multiplexing cell-based assays gives you greater confidence in the parameter you are measuring by putting it in a more biologically relevant context. Multiplexing helps normalize data, improving data accuracy by putting the data into context. Multiplexing also reduces the amount of work and materials needed to perform assays versus making multiple replicates of plates and possibly introducing errors through slight variables in those plates.
References
- Zakowicz, H. et al. (2008) Measuring cell health and viability sequentially by same-well multiplexing using the GloMax®-Multi Detection System. Promega Notes 99, 25–8.
- Bioluminescent Reporters (2011) In: Protocols and Applications Guide, Online Edition, Promega Corporation.
- Niles, A. et al. (2007) Using protease biomarkers to measure viability and cytotoxicity. Cell Notes 19, 16–20.
- CellTiter-Fluor™ Cell Viability Assay Technical Bulletin, TB371, Promega Corporation.
- Apolloni, S. et al. (2010) UDP exerts cytostatic and cytotoxic actions in human neuroblastoma SH-SY5Y cells over-expressing P2Y6 receptor. Neurochem. Intl. 56, 670–8.
- Shultz, S. (2009) Determining the predictive mechanism of toxicity using a single-well multiplexed assay. Promega PubHub.
- Li, W. et al. (2010) Chemical synthesis of 20S-hydroxyvitamin D3, which shows antiproliferative activity. Steroids 75, 926–35.
- He, X. et al. (2010) Resveratrol prevents RNAKL-induced osteoclast differentiation of murine osteosclast progenitor RAW 264.7 cells through inhibition of ROS production. Biochem. Biophys. Res. Comm. 401, 356–62.
- Mulcahy, L.E. et al. (2010) RANKL and OPG activity is regulated by injury size in networks of osteocyte-like cells. Bone 48, 182–8.