Determine Viral-Induced Cytopathic Effect Using a Luminescent Assay
Andrew Niles1, Jim Noah2, Lynn Rasmussen2 and Dan Lazar1
1Promega Corporation; 2Southern Research Institute
Publication Date: December 2013
Abstract
Human history has been shaped, in part, by viral disease. In addition to causing significant morbidity and mortality, viral epidemics and pandemics have influenced societal structure and affected healthcare and hygiene procedures. In modern times, we have battled viral outbreaks by a combination of education, sanitation and vaccination. More recently, antiviral small molecule agents have been developed to complement these practices. Because new viruses are emerging constantly, new therapies that are both safe and potent are needed. Here we describe a new assay which promises to expedite the discovery of new chemical compounds directed against cytolytic viruses, while also providing insight into potential off-target toxicities.
Introduction
Vaccination efforts continue to be a mainstay for viral prophylaxis. However, development of a successful vaccine can be problematic or even elusive due to antigenic drift within the viral pool and variability in the immune responses of the recipients. Other complications occur when experimental vaccines subvert the innate immune system and enhance viral uptake and infectivity rather than preventing infection. Furthermore, the emergence of uncharacterized viruses and speed of transmission in an increasingly globalized society often make normal vaccine development timelines impractical (1) . Therefore, antiviral treatments are needed to bridge this medical need.
The current array of antiviral drugs is usually effective, but prone to viral resistance due to widespread use. Therefore, new more efficient methods of drug discovery are needed to identify novel antiviral compounds from previously unmined libraries (2) . Because many medically important viruses use the lytic life cycle, drug discovery assays that measure host cell health can inversely report viral activity (3) . Traditionally, time-consuming, onerous, morphology-based plaque assays have been used to score cytopathic effect (CPE). More quantitative cell viability chemistries, such as the tetrazolium family of compounds (WST-1, XTT, MTT and MTS) have also been used to define viral infectivity by measuring changes in cellular integrity. However, these assays are poorly suited for high-throughput screening (HTS) due to poor sensitivity or multiple assay steps or both.
We have developed a bioluminescent assay that measures cellular ATP, a tightly regulated molecule in cells, which serves as an excellent surrogate for host cell viability. Here we use four clinically significant viruses and four host cell lines to demonstrate that the Viral ToxGlo™ Assay can determine the relative viral burden in a sample when screening and characterizing antiviral compound potency for drug discovery. Furthermore, the assay could also be easily configured to monitor vaccine development trials by measuring the serum neutralizing potential or immune cell responses directed at the viral target from vaccinated subjects or both.
Understanding and optimizing host cell health and viral infectivity parameters are critical steps for any successful in vitro cytolytic virology effort (4) . Viral infectivity, for instance, is greatly affected by host cell receptor expression and distribution. Efforts to define a susceptible host cell (called viral tropism) and optimal conditions for susceptibility (often linked to cell cycle, passage level and culture conditions) can improve CPE and assay results. Experimentally, this is addressed by titrating the host cell in the presence of a predetermined excess of virus. Titrating host cells without virus is necessary to establish the linear response range of the ATP detection assay.
Once host cell density and linear response range are established, the exposure period for virus/host cell contact needs to be defined. An exposure period that is too short may lead to suboptimal CPE. If the exposure is too long, it may adversely affect the health of uninfected or untreated control cells. Both scenarios would adversely affect the dynamic range of the Viral ToxGlo™ Assay.
In this study, influenza A (subtype H1N1), Venezuelan equine encephalitis virus (VEEV), dengue virus (DENV2) and respiratory syncytial virus (RSV) infectivity were optimized on Madin-Darby canine kidney cells (MDCK), Vero E6 kidney cells, baby hamster kidney cells (BHK-21), and A549 (adenocarcinomic human alveolar) cells, respectively (data not shown).
Results and Discussion
Once infection models are optimized, relative infective virus titers can be determined. For this step, working pools of influenza A, VEEV, dengue virus, and RSV were serially diluted in half-log increments and applied to their respective host cells. Medium only was applied to control host cell wells as a virus-free control. Depending upon the kinetics of CPE (determined during model development), viral titrations were incubated for 72–144 hours. ATP Detection Reagent was prepared and applied in equal volumes to the assay plates (Figure 1). Luminescence was measured and correlated with viral dilutions. The dilutions containing infectious virus demonstrated low cell viability and therefore low luminescence values due to widespread CPE. Conversely, dilutions containing no or limiting virus displayed higher luminescence values. TCID50 values were derived for each virus by determining the dilution of virus that reduced the untreated luminescence signal by 50% (Figure 2, Panels A, B, C and D).
Characterizing On-Target and Off-Target Potencies
The Viral ToxGlo™ technology also can establish antiviral and safety-related potencies. The antiviral agents, ribavirin and interferon β (IFN-β), were diluted in half-log or two-fold serial dilutions, respectively, and applied to host cells. Influenza A, VEEV, dengue virus and RSV were subsequently added to each well of the on-target set of replicate wells at a concentration equal to 100X their respective TCID50. The off-target set of wells received medium without virus to address dose-dependent agent effects. After the appropriate exposure periods, the ATP Detection Reagent was prepared and added to plates as described previously. Luminescence was measured and correlated to antiviral agent concentration for both viral (on-target) and medium (off-target) exposures. The effective potency of the antiviral agent was determined for each combination of virus and agent and expressed as an EC50 value , the concentration of agent that prevented 50% host cell death (Figure 3, Panels A, B, C and D). Off-target toxicity data were plotted on the same graphs and fitted in the case where cytotoxicity was present (ribavirin on BHK-1 cells).
Demonstrating High-Throughput Screening Utility
The final experiment explored the use of the Viral ToxGlo™ Assay for high-throughput screening (HTS). Thirty thousand compounds from the Enamine chemical diversity library were screened as single doses in 384-well plates against three influenza A strains. Doses of H1N1, H3N2 and H5N1 at 100X TCID50 were delivered to MDCK cells in the presence of the Enamine compounds. Infected and uninfected control wells were arrayed on each plate to statistically assess assay robustness. After the cells were incubated for 72 hours with the library compounds, the ATP Detection Reagent was added to the 384-well plates and luminescence measured. Antiviral “hits” were defined as those that demonstrated at least 50% reduction in viral cytotoxicity, with 320 compounds fitting that criterion (data not shown). Lastly, Z´ values were calculated for each plate to evaluate the dynamic range and variation associated with infected and uninfected controls (Figure 4, Panels A, B and C).
Establishing Tissue Culture Infectivity Dose (TCID)
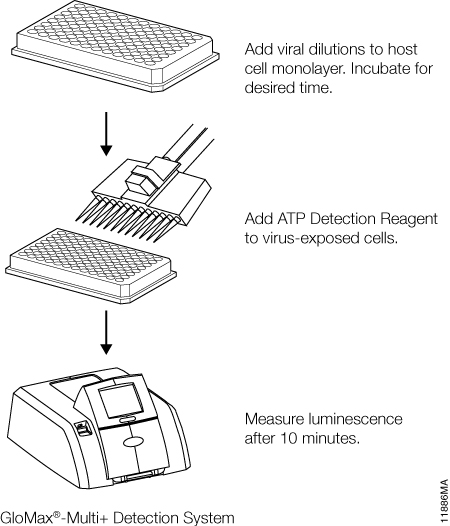
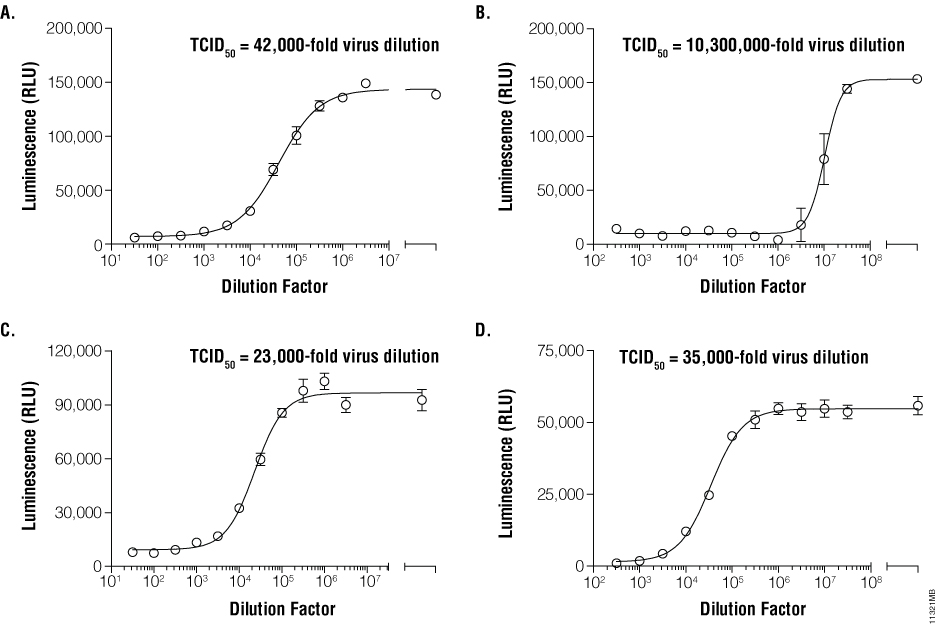
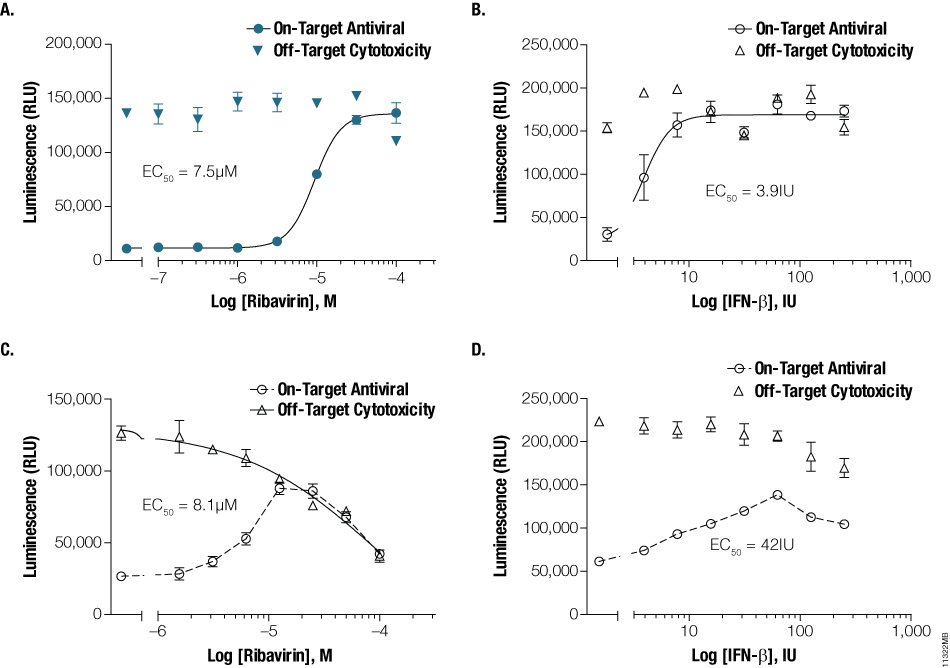
Figure 3. Characterizing on- and off-target antiviral potencies. Ribavirin or IFN-β were serially diluted and applied to MDCK, Vero E6, BHK-21 and A549 monolayers. Influenza (Panel A) or dengue virus (Panel B) at 100X TCID50 was applied to MDCK or BHK-21 containing ribavirin, whereas 100X TCID50 of VEEV (Panel C) or RSV (Panel D) were applied to Vero-E6 or A549 cells containing IFN-β. Culture medium without virus was added to replicate wells containing antivirals to establish off-target, compound-associated cytotoxicity. EC50 values were calculated using GraphPad™ Prism.
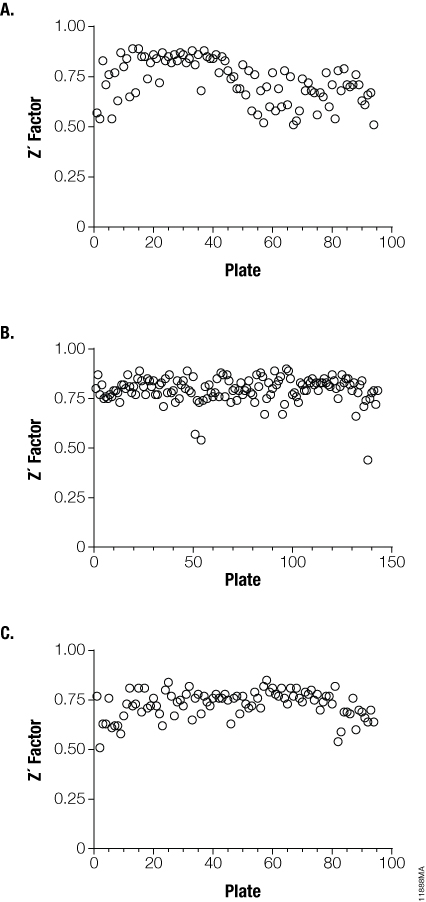
Figure 4. Demonstrating the utility of the Viral ToxGlo™ Assay for HTS screening. Infected and uninfected control wells on influenza H1N1 (Panel A), H3N2 (Panel B) and H5N1 (Panel C) test plates were subjected to Z´ analysis to determine dynamic assay range and variation resident in this screening environment. The data is represented as Z´ factor versus individual plate. Z´ greater than 0.5 are considered to be sufficiently robust for HTS activities.
Summary
The Viral ToxGlo™ Assay is a simple “add-mix-measure” homogeneous assay for monitoring viral-induced cytopathic effect. It offers a highly sensitive, precise and quantitative method of measuring CPE with an easy and fast workflow, an advantage compared to alternate methods. The assay successfully established both TCID50 and on- and off-target potencies for known antiviral compounds against medically relevant viral targets. Furthermore, we demonstrated the use of the Viral ToxGlo™ Assay for HTS, making it possible to use the assay in screening environments and facilitating the search for new, more effective compounds.
References
- Maddry, J.A. et al. (2011) Discovery of novel benzoquinazolinones and thiazoloimidazoles, inhibitors of influenza H5N1 and H1N1 viruses, from a cell-based high-throughput screen. J. Biomol. Screen. 16, 73–78.
- Severson, W.E. et al. (2008) High-throughput screening of a 100,000-compound library for inhibitors of influenza A virus (H3N2). J. Biomol. Screen. 13, 879–887.
- McCoy, M.H. and Wang, E. (2005) Use of electric cell-substrate impedance sensing as a tool for quantifying cytopathic effect in influenza A virus infected MDCK cells in real-time. J. Virol. Methods 130, 157–161.
- Noah, J.W. et al. (2007) A cell-based luminescence assay is effective for high-throughput screening of potential influenza antivirals. Antiviral Res. 73, 50–59.