Strategies for Concordance Testing
National Institute of Standards and Technology, Biochemical Science Division, Gaithersburg, Maryland, USA
Publication Date: 2010
Abstract
Concordance evaluations are important to conduct to determine if there are any allelic dropout or "null alleles" present in a data set. These studies are performed because there are a variety of commercial short tandem repeat (STR) multiplex kits with different configurations of STR markers available to the forensic community. The placement of the markers can vary between kits because the primer sequences were designed to amplify different polymerase chain reaction (PCR) product sizes. When multiple primer sets are used, there is concern that allele dropout may occur due to primer-binding-site mutations that affect one set of primers but not another. These null alleles become evident only when data sets are compared. Multiple concordance studies have been performed at NIST with a standard sample set using various STR multiplex kits, including Identifiler®, MiniFiler™, PowerPlex® 16, ESX 17 and ESI 17 Systems, and in-house assays. Various discordant results have been identified using concordance software developed at NIST, confirmed by DNA sequencing and reported to the forensic community on the null allele web page of STRBase.
Introduction
There are multiple commercial STR multiplex kits for DNA typing available to the forensic community. Most of these STR kits simultaneously amplify the same markers or subsets of the same markers for genotyping. There are two different strategies when designing kit configurations with many of the same STR loci. The first is to use different PCR primer sequences that place the loci in different positions within the separate kits. This allows concordance testing to be performed to compare results between kits and thus detect allelic dropout or “null alleles” within the data due to primer-binding-site mutations. It is then possible to redesign primer sequences to avoid these mutations, resulting in the correct genotype.
The second strategy is to keep the PCR primer sequences the same and use mobility-modifying non-nucleotide linkers, or “mobility modifiers”, allowing the markers to be located in different positions. These mobility modifiers permit amplified alleles from one member of a pair of closely spaced STR loci to be shifted relative to the other, preventing overlapping size ranges (1) . By using this technology in STR multiplex development, primer sequences can be maintained to amplify STR loci with optimal interlocus spacing within the various color channels. This enables comparison of data sets between kits with overlapping markers without the concern of discordant results. There are advantages and disadvantages of using either strategy, which are summarized in Table 1.
 Table 1. The Advantages and Disadvantages of Two Strategies for the Multiplex Kit Design and Placement of Loci.
Table 1. The Advantages and Disadvantages of Two Strategies for the Multiplex Kit Design and Placement of Loci. There are two primary vendors of STR multiplex kits: Applied Biosystems (Foster City, CA) and Promega Corporation (Madison, WI). The kits from these two companies have different configurations of many of the same STR markers. To enable this difference in marker placement, primer sequences were designed to amplify PCR products with sizes that vary between kits. In addition to differences in kits distributed by different vendors, Promega has redesigned or changed primer sequences for many loci between PowerPlex® versions, including PowerPlex® 16, ESX 17 and ESI 17. The amplicon sizes in base pairs (bp) are different for many overlapping loci in these multiplex PCR systems. The size differences were determined for each locus in PowerPlex® 16 and ESI 17 and Identifiler® (Applied Biosystems) kits relative to the PowerPlex® ESX 17 System and are listed in Table 2 (2) .
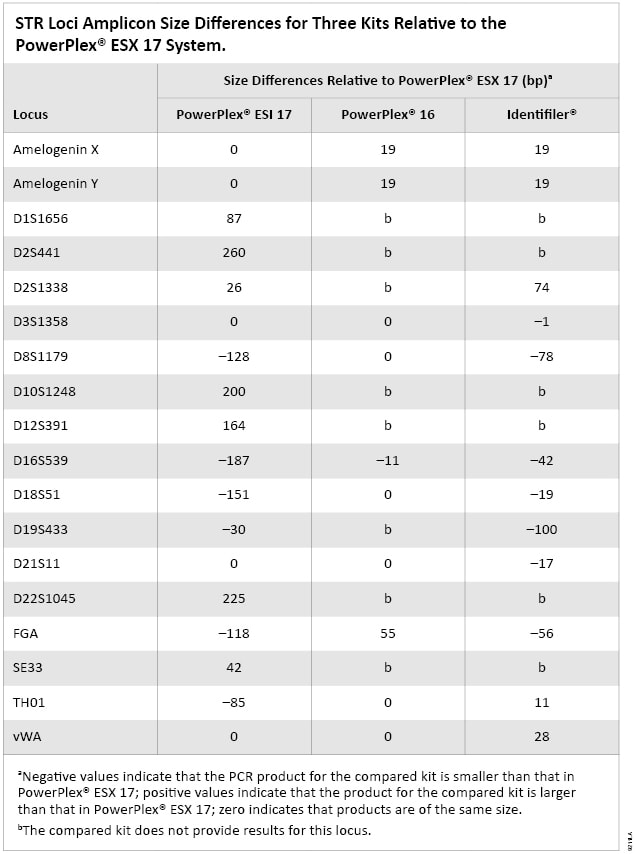 Table 2. STR Loci Amplicon Size Difference for Three Kits Relative to the PowerPlex® ESX 17 System.
Table 2. STR Loci Amplicon Size Difference for Three Kits Relative to the PowerPlex® ESX 17 System. In contrast, Applied Biosystems alters the configurations of their multiplex PCR kits by using mobility modifiers and preserving primer sequences. One exception is the MiniFiler™ kit in which primers were redesigned as compared to the Identifiler® kit to allow normally large amplicons to be made smaller. A concordance evaluation was conducted at NIST and reported on previously (3) .
The U.S. national DNA database stores only genotype information for a sample, relying on input from different STR kits such as PowerPlex® 16 and Identifiler®. The use of the same kit or kits with unchanging primer sequences makes concordance studies unnecessary since the data sets will be the same and null alleles would be undetected (Table 1). However, when other kits are used with different primers, null alleles could potentially result in discordant results being entered into the database. To overcome this problem, the Combined DNA Index System (CODIS) software permits moderate- and low-stringency matches when searching an evidence sample against the DNA database of convicted offender profiles (1) . However if these samples were tested with the same kit, they would provide the same result; thus, a primer-binding-site mutation would not be a problem in this case.
Even though concordance studies are important to find null alleles between data sets, discordant results rarely occur for most primer sets. At NIST, concordance evaluations have been performed for over 150,000 allele comparisons. In the MiniFiler™ concordance study (3) , 99.7% full concordance was observed, and in the PowerPlex® ESX 17/ESI 17 study (2) , full concordance was seen for >99.8% in all comparisons performed.
Concordance Evaluations—The Four “S”s of Concordance
Concordance studies are necessary when comparing data sets resulting from STR multiplex kits that have varying configurations of the same loci. These markers can be in different positions because they have alternative primer sequences. Because of this, there is the potential for allelic dropout or a “null allele” if a primer-binding-site mutation affects one of the primer pairs. The purpose of concordance testing is illustrated in Figure 1. The use of nonoverlapping primers permits the detection of allele dropout. Null alleles are a concern because this could result in a false-negative or incorrect exclusion of two samples that come from a common source (only if different PCR primers are used). A base pair change in the DNA template at the PCR-primer-binding region can disrupt primer hybridization and result in a failure to amplify and detect an existing allele (1) .
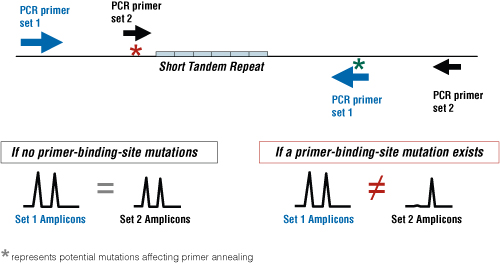 Figure 1. The purpose of concordance evaluations is to compare resultant data sets from different primer pairs (nonoverlapping) to detect allelic dropout or null alleles.
Figure 1. The purpose of concordance evaluations is to compare resultant data sets from different primer pairs (nonoverlapping) to detect allelic dropout or null alleles. To test for concordance between data sets, our strategy at NIST is to use standard samples, software, sequencing and STRBase—what we might term the four “S”s of concordance. Ultimately, the information can be used by kit manufacturers when designing new STR multiplexes to either add an extra (degenerate) primer or redesign primers away from primer-binding-site mutations in the final kit configurations. Some kit manufacturers decide not to change the primer sequences and rely simply on the documentation or publication of the reported null alleles (3) as illustrated in Figure 2.
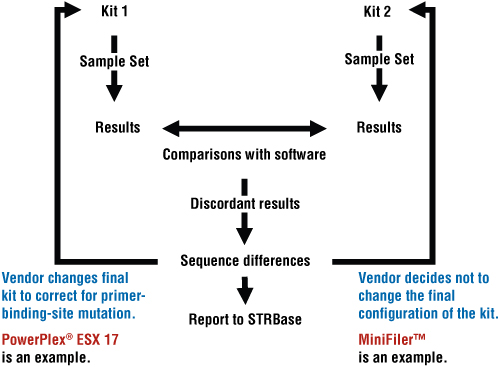 Figure 2. Summary of the steps used in concordance testing and how they affect STR multiplex development.
Figure 2. Summary of the steps used in concordance testing and how they affect STR multiplex development. Standard Sample Set
Concordance testing is a multistep process to compare the results of a standard sample set when run with different STR multiplex kits. The first step is to determine which samples will be used for concordance evaluations. At NIST, there are several in-house U.S. population samples (~1450 total) that have been run with a variety of STR multiplex kits, including MiniFiler™ (3) and Identifiler® (4) from Applied Biosystems, as well as PowerPlex® 16, ESX 17 and ESI 17 (5) from Promega. Many in-house assays also have been tested, including the nonCODIS (NC) miniSTR assays (6) (7) and NIST 26plex (8) . A summary of the comparisons performed and allele discordance observed with the NIST data sets is shown in Figure 3. Genotyping data from these studies are or will be available at: www.cstl.nist.gov/biotech/strbase/NISTpop.htm
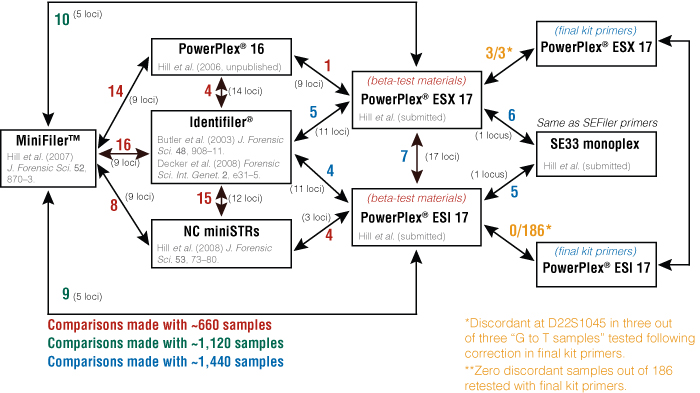 Figure 3. Summary of all concordance evaluations performed at NIST, including allele discordance observed.
Figure 3. Summary of all concordance evaluations performed at NIST, including allele discordance observed.
The number of disconcordant results observed during each comparison is indicated.
Concordance Software
Comparisons of data sets are made possible with software developed at NIST using Excel® (Microsoft®, Redmond, WA) macros. These programs can be found at: www.cstl.nist.gov/biotech/strbase/software.htm. There are two programs of note that are valuable for concordance evaluations. The first is called STR_ConvertFormats, which converts output files from GeneMapper® ID (Applied Biosystems) to a universal input file. The primary concordance program used in our studies is called STR_MatchSamples and is an Excel®-based tool developed to aid comparison of STR genotypes from two or more data sets. The “Exact Match” function creates a list of all samples that are fully concordant at all loci between the samples being compared. The remaining discordant samples are highlighted for further review.
DNA Sequencing
With every discordant result identified in the NIST concordance software, DNA sequencing was performed to validate the result and determine the exact cause of the allele dropout. Most of the examples were due to primer-binding-site mutations; however, in some cases there was a difference in allele calls between two primer sets due to an insertion or deletion in the DNA template. This occurred in three samples for the CSF1PO, D7S820 and SE33 loci. Differences detected between discordant loci within the kits compared over the past few years are shown in Table 3 (3) (5) . Information on variant and null alleles that have been sequenced at NIST can be found at STRBase at: www.cstl.nist.gov/biotech/strbase/STRseq.htm
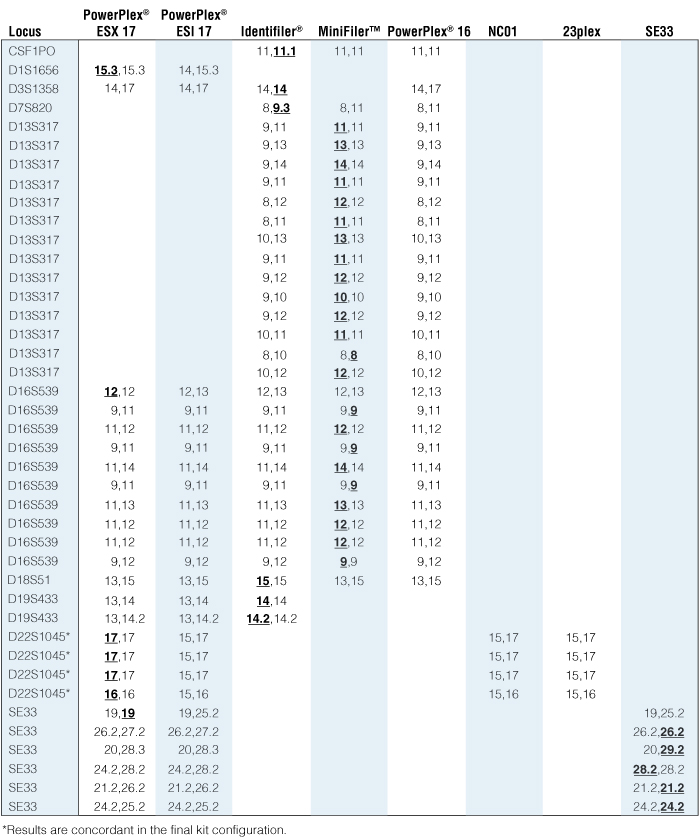 Table 3. Discordant results observed (41 total) in over 150,000 concordance comparisons performed at NIST.
Table 3. Discordant results observed (41 total) in over 150,000 concordance comparisons performed at NIST.
The underlined, bolded alleles exhibited allelic dropout or an insertion or deletion. Not all discordancies are within one multiplex kit. Instead, they span four kits tested (PowerPlex® ESX 17, Identifiler®, MiniFiler™ and SE33).
An example of discordance due to a null allele was observed at the D19S433 locus. The Identifiler® genotype was a 14,14, and the genotype for both PowerPlex® ESX 17 and ESI 17 was a 13,14, indicating allelic dropout in Identifiler® (Figure 4, Panel A). All three kits have different primer sequences for this marker (Table 2); thus, the null allele is most likely due to a primer-binding-site mutation in one of the Identifiler® primers. DNA sequencing was performed on these samples, and a G→A single nucleotide polymorphism (SNP) was observed 32bp downstream from the repeat that affected reverse primer binding with Identifiler® (Figure 4, Panel B). There is a higher frequency of this mutation for D19S433 in Asian populations than others, which was reported previously (9) . Interestingly, all null alleles found in the NIST data set for D19S433 were with Asian samples.
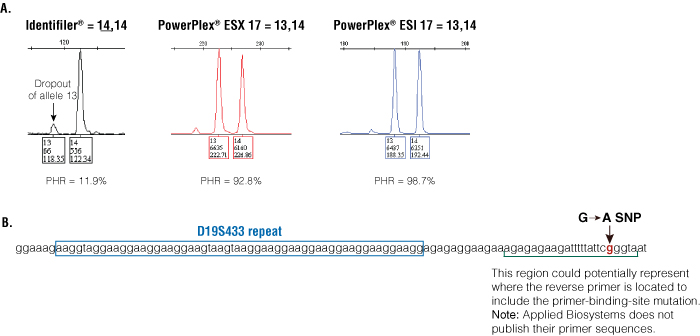 Figure 4. An example of discordance with the D19S433 between three kits.
Figure 4. An example of discordance with the D19S433 between three kits.
Panel A. Peak heights and peak height ratios (PHR) for the discordant locus are shown. Panel B. DNA sequence of the D19S433 template to illustrate the G→A SNP 32bp downstream from the repeat in the reverse primer-binding site.
STRBase Web Site on Null Alleles
Once null alleles and discordant genotypes are verified through DNA sequencing, these results can be reported on the NIST STRBase web site. Laboratories may contribute confirmed null alleles from their sample sets through a link on this page. It also contains a section called “Results from Concordance Studies”, which summarizes all discordant results that have been identified at NIST and submitted from the forensic community. In addition, this null allele page on STRBase has links to variant alleles, tri-allelic patterns and a comprehensive listing of relevant literature. This web site can serve as a useful tool to validate discordant findings when performing concordance studies.
Conclusions
Concordance evaluations are useful to identify null alleles in data sets. They are especially important when developing new STR multiplex kits when primer redesign is involved. If allelic dropout is seen in one data set as compared to another for a certain marker and DNA sequencing confirms a primer-binding-site mutation, there are ways of improving the final kit configuration by adding an extra (degenerate) primer or redesigning the primer to move a primer away from a high-frequency mutation before a kit is released to the forensic community. In some cases, an additional PCR primer can be added to the assay to hybridize properly to the alternative allele when it exists in a sample. In other instances, primer redesign is the best course of action to overcome allele dropout issues. Sometimes in occurrences of rare mutations, no improvements are made and the null alleles are carefully documented for future reference. In all situations, concordance studies are a reliable way to determine if a null allele is present to avoid misinterpretation of data.
Acknowledgements
This work was funded in part by the National Institute of Justice (NIJ) through an interagency agreement 2008-DN-R-121 with the NIST Office of Law Enforcement Standards. Points of view in this document are those of the authors and do not necessarily represent the official position or policies of the U.S. Department of Justice. Certain commercial equipment, instruments and materials are identified in order to specify experimental procedures as completely as possible. In no case does such identification imply a recommendation or endorsement by the National Institute of Standards and Technology nor does it imply that any of the materials, instruments or equipment identified are necessarily the best available for the purpose.
Article References
- Butler, J.M. (2010) Fundamentals of Forensic DNA Typing, Elsevier Academic Press
- Hill, C.R. et al. (2011) Concordance and population studies along with stutter and peak height ratio analysis for the PowerPlex® ESX 17 and ESI 17 Systems. Forensic Sci. Int. Genet. 5, 269–75.
- Hill, C.R. et al. (2007) Concordance study between the AmpFlSTR® MiniFiler™ PCR amplification kit and conventional STR typing kits. J. Forensic Sci. 52, 870–3.
- Butler, J.M. et al. (2003) Allele frequencies for 15 autosomal STR loci on U.S. Caucasian, African American, and Hispanic populations. J. Forensic Sci. 48, 908–11.
- Coble, M.D. and Butler, J.M. (2005) Characterization of new miniSTR loci to aid analysis of degraded DNA. J. Forensic Sci. 50, 43–53.
- Hill, C.R. et al. (2008) Characterization of 26 miniSTR loci for improved analysis of degraded DNA samples. J. Forensic Sci. 53, 73–80.
- Hill, C.R., Butler, J.M and Vallone, P.M. (2009) A 26plex autosomal STR assay to aid human identity testing. J. Forensic Sci. 54, 1008–15.
- Mizuno, N. et al. (2008) A D19S433 primer binding site mutation and the frequency in Japanese of the silent allele it causes. J. Forensic Sci. 53, 1068–73.
How to Cite This Article
Scientific Style and Format, 7th edition, 2006
Hill, C.R. et al. Strategies for Concordance Testing. [Internet] 2010. [cited: year, month, date]. Available from: https://www.promega.com/resources/profiles-in-dna/2010/strategies-for-concordance-testing/
American Medical Association, Manual of Style, 10th edition, 2007
Hill, C.R. et al. Strategies for Concordance Testing. Promega Corporation Web site. https://www.promega.com/resources/profiles-in-dna/2010/strategies-for-concordance-testing/ Updated 2010. Accessed Month Day, Year.
Contribution of an article to Profiles in DNA does not constitute an endorsement of Promega products.
Products may be covered by pending or issued patents or may have certain limitations. More information.
All prices and specifications are subject to change without prior notice.
Product claims are subject to change. Please contact Promega Technical Services or access the Promega online catalog for the most up-to-date information on Promega products.