Transfection
Protocols for transfection and information on optimization
This guide provides an overview of transfection methods and provides example protocols and guidance for choosing a transfection reagent. Visit the Transfection Reagents product page to view ordering information for Promega Transfection Reagents.
An Introduction to Transfection
Transfection is the process of introducing nucleic acids into eukaryotic cells by nonviral methods. Using various chemical or physical methods, this gene transfer technology enables the study of gene function and protein expression in a cellular environment. The development of reporter gene systems and selection methods for stable maintenance and expression of transfected DNA have greatly expanded the applications for transfection. Assay-based reporter technology, together with the availability of a wide array of transfection reagents, provides the foundation to study mammalian promoter and enhancer sequences, trans-acting proteins such as transcription factors, mRNA processing, protein:protein interactions, translation and recombination events (Groskreutz and Schenborn, 1997).
Transfection overcomes the inherent challenge of introducing negatively charged molecules (e.g., phosphate backbones of DNA and RNA) into cells with a negatively charged membrane. Chemicals like calcium phosphate and diethylaminoethyl (DEAE)‐dextran or cationic lipid-based reagents coat the DNA, neutralizing or even imparting an overall positive charge to the molecule (Figure 1). This process makes it easier for the DNA:transfection reagent complex to cross the membrane, especially for lipids that have a “fusogenic” component, which enhances fusion with the lipid bilayer. Physical methods like microinjection or electroporation simply punch through the membrane and introduce DNA directly into the cytoplasm. Each of these transfection technologies is discussed in the following sections.
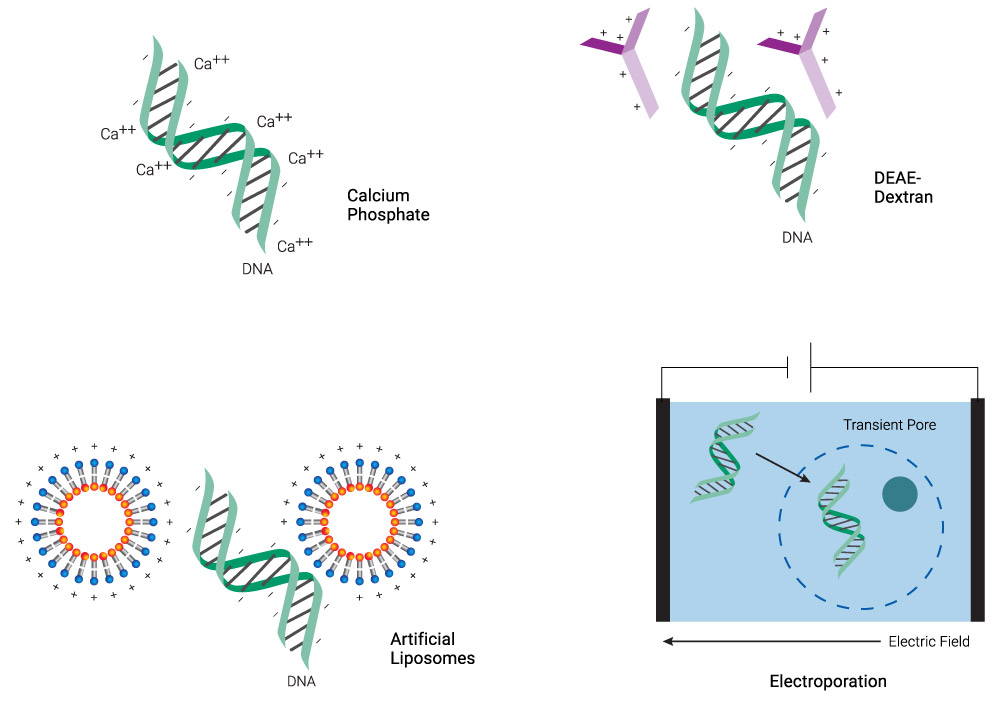
Figure 1. Schematic representation of common transfection methods. Chemical methods neutralize the negative charge of DNA, facilitating its uptake. Lipid-based reagents can also coat DNA while forming micelles. Electroporation makes the membrane more permeable transiently, allowing DNA to enter the cell.
This chapter covers general information on transfection techniques and considerations for transfection efficiency and optimization. In addition, we discuss various transfection agents and general protocols for transfection, with specific examples using our transfection reagents. Finally, we review stable transfection and outline a protocol using drug selection.
Chemical Reagents
DEAE-dextran, a cationic polymer, was one of the first chemical reagents used to transfect cultured mammalian cells (Vaheri and Pagano, 1965; McCutchan and Pagano, 1968). An excess of positive charge, contributed by the polymer in the DNA:polymer complex, allows the complex to come into closer association with the negatively charged cell membrane. Uptake of the complex is presumably by endocytosis. This method successfully delivers nucleic acids into cells for transient expression; that is, for short-term expression studies of a few days. However, DEAE-dextran is not generally useful for stable or long-term transfection studies that rely upon integration of transferred DNA into the chromosome. Other synthetic cationic polymers have been used to transfer DNA into cells, including polybrene (Kawai and Nishizawa, 1984), polyethylenimine (Boussif et al. 1995) and dendrimers (Haensler and Szoka, 1993; Kukowska-Latallo et al. 1996).
Calcium phosphate co-precipitation as a transfection method was introduced in the early 1970s (Graham and van der Eb, 1973). The method is still popular because the components are easily available and inexpensive, the protocol is easy to use, and it is effective with many different types of cultured cells. The protocol involves mixing DNA with calcium chloride, adding this mixture in a controlled manner to a buffered saline/phosphate solution and incubating the mixture at room temperature. The controlled addition generates a precipitate that is dispersed onto the cultured cells. The precipitate is taken up by cells via endocytosis or phagocytosis. Calcium phosphate transfection is routinely used for both transient and stable transfection of a variety of cell types. However, calcium phosphate co-precipitation is prone to variability and is not suited for in vivo gene transfer into whole animals. In addition, small pH changes (± 0.1) can compromise transfection efficiency (Felgner, 1990).
Due to their shortcomings, chemical reagents have been largely superseded by synthetic lipid-based reagents.
Cationic Lipids
The term “liposome” refers to lipid bilayers that form colloidal particles in an aqueous medium (Sessa and Weissmann, 1968). Artificial liposomes were first used to deliver DNA into cells in 1980 (Fraley et al. 1980). The next advance in liposomal vehicles was the development of synthetic cationic lipids (Felgner et al. 1987). The cationic head group of the lipid compound associates with negatively charged phosphates on the nucleic acid. Liposome-mediated delivery offers advantages such as relatively high efficiency of gene transfer, the ability to transfect certain cell types that are resistant to calcium phosphate or DEAE-dextran, in vitro and in vivo applications, successful delivery of DNA of all sizes from oligonucleotides to yeast artificial chromosomes, delivery of RNA, and delivery of protein (reviewed in Kim and Eberwine, 2010; Stewart et al. 2016). Cells transfected by liposome techniques can be used for transient expression studies and long-term experiments that rely on integration of DNA into the chromosome or episomal maintenance. Unlike DEAE-dextran or calcium phosphate chemical methods, liposome-mediated nucleic acid delivery can be used for in vivo transfer of DNA and RNA into animals and humans (Felgner et al. 1995).
A lipid with overall net positive charge at physiological pH is the most common component of liposomes developed for gene delivery (Figure 2). Often, the cationic lipid is mixed with a neutral lipid such as L‑dioleoyl phosphatidylethanolamine (DOPE; Figure 3), which can enhance the gene transfer ability. The cationic portion of the lipid molecule associates with negatively charged nucleic acids, resulting in compaction of the nucleic acid in a liposome/nucleic acid complex (Kabanov and Kabanov, 1995; Labat-Moleur et al. 1996). For cultured cells, the overall net positive charge of the liposome/nucleic acid complex generally results in higher transfer efficiencies, presumably because it allows closer association of the complex with the negatively charged cell membrane. Entry of the liposome complex into the cell may occur by endocytosis or fusion with the plasma membrane via the lipid moieties of the liposome (Gao and Huang, 1995). Following cellular internalization, the complexes appear in the endosomes and later in the nucleus. It is unclear how the nucleic acids are released from the endosomes and lysosomes and traverse the nuclear membrane. DOPE is considered a “fusogenic” lipid (Farhood et al. 1995), and its role may be to release these complexes from endosomes as well as to facilitate fusion of the outer cell membrane with liposome/nucleic acid complexes. While transfected DNA will need to enter the nucleus, the cytoplasm is the site of action for RNA, protein or antisense oligonucleotides delivered via liposomes.
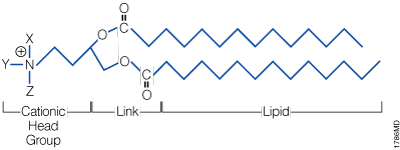
Figure 2. The general structure of a synthetic cationic lipid. X, Y and Z represent a number of possible chemical moieties, which can differ, depending on the specific lipid.
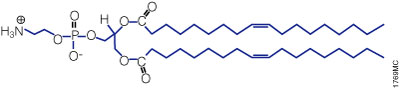
Figure 3. Structure of the neutral lipid DOPE. The ViaFect™ Transfection Reagent (Cat.# E4981) is a cationic formulation designed to transfect DNA into a wide variety of cell lines with high efficiency and low toxicity. It is especially suited for cell types that are traditionally difficult to transfect.
Nonliposomal Reagents
Although liposomal transfection reagents have a wide range of applications, they may not be efficient in all cell types. Nonliposomal transfection reagents offer an alternative to liposome-mediated transfection methods. Such reagents include polymers that are capable of forming complexes with DNA or RNA, and other types of lipids that can form micelles in aqueous solutions. Like liposomal transfection reagents, they offer low toxicity, reproducible results, high efficiency and are suitable for a variety of cell types. Lipid nanoparticles represent an extension of this transfection method that is especially suited for the delivery of small-molecule drugs in clinical research and therapeutic applications (reviewed in Cullis and Hope, 2017).
Examples of nonliposomal transfection reagents include FuGENE® HD Transfection Reagent (Cat.# E2311), suitable for cell types that are difficult to transfect; FuGENE® 6 Transfection Reagent (Cat.# E2691), suitable for a wide variety of cell lines with high efficiency and low toxicity; and FuGENE 4K Transfection Reagent (Cat.# E5911), a high-efficiency reagent for both challenging and routine cell lines. You should determine the best transfection reagent and conditions for a particular cell type empirically, because inherent properties of the cell influence the success of any specific transfection method.
Physical Methods
Physical methods for gene transfer were developed beginning in the early 1980s. Direct microinjection into cultured cells or nuclei is an effective although laborious technique to deliver nucleic acids into cells using a fine needle (Cappechi, 1980). This method was widely used to introduce foreign DNA into Xenopus oocytes and Drosophila embryos. Although relatively inefficient, microinjection is still used to transfer DNA into embryonic stem cells that are used to produce transgenic organisms (Bockamp et al. 2002), particularly for genetic research in farm animals (Wall, 2001). Recently, microinjection has regained popularity in gene-editing applications. For example, it has been used to deliver CRISPR-Cas9 DNA into zygote pronuclei to create knockout pigs (Chuang et al. 2017).
Electroporation was first reported for gene transfer studies in mouse cells (Wong and Neumann, 1982). The mechanism is based on the use of an electrical pulse to perturb the cell membrane and form transient pores that allow passage of nucleic acids into the cell (Shigekawa and Dower, 1988). The technique requires optimization of pulse duration and strength for each type of cell used. Electroporation often requires more cells than chemical methods because it results in substantial cell death, and extensive optimization often is required to balance transfection efficiency with cell viability. Modern instrumentation allows nucleic acid delivery to the nucleus and successful transfer of DNA and RNA into primary and stem cells. Electroporation has also been used in combination with physical constriction of cells in microfluidic devices (Ding et al. 2017) for rapid gene expression in a high-throughput format.
Another physical method of transfection is biolistic particle delivery, also known as particle bombardment. This method relies upon high-velocity delivery of nucleic acids on microprojectiles to recipient cells by membrane penetration (Ye et al. 1990). It has been used to deliver nucleic acid to cultured cells as well as to cells in vivo (Klein et al. 1987; Burkholder et al. 1993; Ogura et al. 2005). Initially, the high cost of particle bombardment instruments precluded their use in research applications and the method was primarily used in agricultural genetics. However, recent developments in nanoparticle technology have opened up new applications. Nanoparticles are synthetic materials in which at least one of the particle dimensions are less than 100nm. Due to their small size, nanoparticles are able to pass through cell membranes with high efficiency, and they can bind DNA and RNA through covalent or noncovalent interactions (An and Jin, 2012). These properties have been exploited for creating transgenic animals, as an alternative to the use of pronuclear microinjection or viral vectors (Dziegiel, 2016). A variation of this method is using iron oxide nanoparticles for gene delivery in the presence of strong magnetic fields, or “magnetofection” (Dziegiel, 2016).
General Considerations
Reagent Selection
No transfection reagent is ideal for use in all cell types and under all conditions, and optimization of experimental conditions is required with any reagent. With so many different methods of gene transfer, how do you choose the right transfection reagent or technique for your needs? Although traditional chemical methods for transfection are still in use, lipid-based reagents have become popular due to their ease of use and applicability to a variety of cell types. Lipofectamine® reagent (Thermo Fisher) and its newer derivatives are widely used for both transient and stable transfection experiments. FuGENE® Transfection Reagents are also popular due to their ease of use and compatibility with a variety of cell lines. FuGENE® 4K, FuGENE® HD and FuGENE® 6 products provide ideal conditions for transfecting DNA into a many different cell lines.
The performance of frequently used transfection reagents in many common cell lines is provided in Table 1. Transfection was assessed through expression of firefly luciferase. The best condition (highest RLUs) was chosen for each transfection reagent. For each cell line, the maximum RLUs were set at 100% for one reagent and the other three were compared relative to that reading. Cells were also assayed for cell viability and compared with a no-transfection control for each cell line. Transfection efficiencies lower than 50% were not reported in this table. ViaFect™ Reagent offers performance similar to or better than that of Lipofectamine® 2000 reagent, and it may be suitable for some cell lines where other lipid-based reagents result in lower transfection efficiencies (Table 1). In cases where lipid-based reagents are unsuitable, electroporation may offer an alternative; however, a larger number of cells will be required due to reduced cell viability.
Table 1. Comparison of Transfection Reagents.
| Cell Line | FuGENE® 4K | FuGENE® 6 | FuGENE® HD | ViaFect™ Reagent | Competitor L2K | ||||||
| EXP1 | VIA1 | EXP2 | VIA2 | EXP2 | VIA2 | EXP2 | VIA2 | EXP1,2 | VIA1,2 | ||
| A549 | ++ | +++ | ++ | ++ | +++ | +++ | +++ | +++ | |||
| C2C12 | +++ | +++ | +++ | ||||||||
| CHO | +++ | ++ | ++ | +++ | +++ | ++ | ++ | ++ | |||
| COS7 | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | |||
| H9C2 | +++ | +++ | ++ | +++ | |||||||
| HCT116 | +++ | +++ | +++ | ++ | +++ | +++ | ++ | ++ | |||
| HEK 293 | +++ | ++ | ++ | ++ | +++ | +++ | |||||
| HeLa | ++ | +++ | +++ | +++ | ++ | +++ | |||||
| HepG2 | ++ | +++ | +++ | ++ | |||||||
| HT-29 | +++ | ++ | +++ | +++ | +++ | +++ | |||||
| Huh7 | +++ | ++ | ++ | ++ | |||||||
| Jurkat | +++ | +++ | +++ | +++ | ++ | +++ | |||||
| K562 | +++ | ++ | +++ | +++ | |||||||
| LNCaP | ++ | +++ | +++ | +++ | |||||||
| MCF7 | ++ | +++ | +++ | +++ | +++ | +++ | |||||
| NIH 3T3 | +++ | +++ | ++ | +++ | +++ | +++ | |||||
| PC-12 | ++ | +++ | +++ | +++ | +++ | +++ | |||||
| PC-3 | +++ | +++ | +++ | +++ | |||||||
| RAW 264.7 | +++ | +++ | +++ | ++ | +++ | ++ | ++ | ++ | |||
| U2OS | +++ | +++ | ++ | +++ | +++ | +++ | |||||
| +++ | >80% of max RLUs; >80% of cells were viable | ++ | >50% to <80% max RLUs; >50% to <80% of cells were viable |
| EXP, transfection efficiency based on flow cytometry cell counts1 or expression of the transfected luciferase gene2; VIA, cell viability assessed by propidium iodide dye1 or the CellTiter-Fluor™ Cell Viability Assay compared to a no-transfection control2 | |||
Any time a new parameter, like a new cell line, is introduced, you will need to determine the optimal conditions for transfection. This process may involve choosing a new transfection reagent. For example, one reagent may work well with HEK-293 cells, but another reagent may be a better choice when using HepG2 cells. The Transfection Assistant offers a drop-down menu to select cell lines and either FuGENE® HD or FuGENE® 6 Transfection Reagent for transfection conditions. The conditions should be considered only guidelines since you may need to optimize the transfection conditions for your specific application. See Optimization of Transfection Efficiency and General Transfection Protocol for details.
Transient Expression versus Stable Transfection
Another parameter to consider is the time frame of the experiment you wish to conduct. For instance, determining which promoter deletion constructs can still function as a promoter can be accomplished with a transient transfection experiment, while establishing stable expression of an exogenously introduced gene construct will require a longer-term experiment. Stable transfection results in integration of the transfected gene into the host cell genome, and it is useful for studying long-term effects of gene expression or for creating cell lines with novel characteristics for further experiments, e.g., screening potential drug candidates in discovery experiments.
Transient Expression
Cells are typically harvested 24–72 hours after transfection for studies designed to analyze transient expression of transfected genes. The optimal time interval depends on the cell type, research goals and specific expression characteristics of the transferred gene. Analysis of gene products may require isolating RNA or protein for enzymatic activity assays or immunoassays. The method used for harvesting cells will depend on the end product assayed. For example, expression of the firefly luciferase gene in the pGL4.10[luc2] Vector (Cat.# E6651) is generally assayed 24–48 hours after transfection, whereas the pGL4.12[luc2CP] Vector (Cat.# E6671) with its protein degradation sequences can be assayed in a shorter time frame (e.g., 3–12 hours), depending on the research goals and the time it takes for the reporter gene to reach steady-state expression.
When performing a transient transfection, you can choose between a standard or reverse transfection protocol (see Figure 4). In a standard transfection protocol, the cells are plated on day 1, transfected on day 2 and assayed on day 3 or 4. In a reverse transfection protocol, cells are added directly to a plate containing the transfection reagent/DNA mix and assayed on day 2 or 3. Because the cells are added directly to the DNA, this process reduces the experimental time by one day and allows for high-throughput transfection of DNA in a plate or microarray format. For more information on reverse transfection, see a protocol using FuGENE® 6 and FuGENE® HD Reagents.

Figure 4. Overview of various transfection methods.
Stable Transfection
The goal of stable, long-term transfection is to isolate and propagate individual clones containing transfected DNA that has integrated into the cellular genome. Distinguishing nontransfected cells from those that have taken up exogenous DNA involves selective screening. This screening can be accomplished by drug selection when an appropriate drug-resistance marker is included in the transfected DNA. Alternatively, morphological transformation can be used as a selectable trait in certain cases. For example, bovine papilloma virus vectors produce a morphological change in transfected mouse CI127 cells (Sarver et al. 1981).
Before using a particular drug for selection purposes, you will need to determine the amount of drug necessary to kill nontransfected cells. This amount may vary greatly among cell types and should be optimized for each new cell line being transfected.
When drug selection is used, cells are maintained in nonselective medium for 1–2 days after transfection, then replated in selective medium containing the drug. The use of selective medium is continued for 2–3 weeks, with frequent changes of medium to eliminate dead cells and debris, until distinct colonies can be visualized. Individual colonies can be isolated by cloning cylinders, selected and transferred to multiwell plates for further propagation in the presence of selective medium. Individual cells that survive the drug treatment expand into clonal groups that can be individually propagated and characterized. For a protocol to select transfected cells by antibiotics, see Stable Transfection.
Several different drug-selection markers are commonly used for long-term transfection studies. For example, cells transfected with recombinant vectors containing the bacterial gene for neomycin phosphotransferase [e.g., pCI-neo Mammalian Expression Vector (Cat.# E1841)] can be selected for stable transformation in the presence of the neomycin analog G-418 (Cat.# V8091). Similarly, expression of the hygromycin B phosphotransferase gene from the transfected vector [e.g., pGL4.14(luc2/Hygro) Vector (Cat.# E6691)] will confer resistance to the drug hygromycin B.
An alternative strategy is to use a vector carrying an essential gene that is defective in a given cell line. For example, CHO cells deficient in dihydrofolate reductase (DHFR) gene expression do not survive without added nucleosides. However, these cells, when stably transfected with DNA expressing the DHFR gene, will synthesize the required nucleosides and survive. An additional advantage of using DHFR as a marker is that gene amplification of DHFR and associated transfected DNA occurs when cells are exposed to increasing doses of methotrexate, resulting in multiple copies of the plasmid in the transfected cell (Schimke, 1988).
Type of Molecule Transfected
Plasmid DNA is most commonly transfected into cells, but other macromolecules can be transferred as well. For example, DNA oligonucleotides, small interfering RNA (siRNA), proteins, and ribonucleoprotein complexes (RNPs) have been successfully introduced into cells via transfection methods (reviewed in Stewart et al. 2016). However, conditions that work for plasmid DNA transfer will likely need to be optimized when using other macromolecules. In all cases, the agent transfected needs to be of high quality and relatively pure. Nucleic acids should be free of proteins, other contaminating nucleic acids and chemicals (e.g., salts from oligo synthesis). Protein should be pure and in a solvent that is not detrimental to cell health.
Cell Type
Selecting the cell type is one decision that is vital for a successful transfection experiment. Due to the variability in cell responses that may occur with different reagents or methods, choosing the appropriate cell type when designing an experiment can help maximize transfection success.
One critical consideration is that the activity of certain promoters can be modified in different cell types. Primary cultures can represent natural tissue but are more difficult to maintain over time in culture. In contrast, using routine and well-established cell lines that able to be maintained over a long period of time can result in genetic changes that may differ from the model being mimicked. Considering the promoter, and how it will affect successful expression, is an important decision as well as the understanding the parameters that affect the in vivo situation being investigated.
Expanding and maintaining cells for use in transfection experiments can prove quite challenging due to the large amount of effort and time needed. The easy-to-use TransFectNow™ HEK293 Cells can be directly transfected by simply thawing and diluting saves the time required to both expand and maintain cultured cells.
Assays for Transfection
After cells are transfected, how will you determine success? Plasmids containing reporter genes can be used to easily monitor transfection efficiencies and expression levels in the cells. An ideal reporter gene product is one that is unique to the cell, can be expressed from plasmid DNA and can be assayed conveniently. Generally, reporter gene assays are performed 1–3 days after transfection; the optimal time should be determined empirically. A functional test for the protein of interest, such as an enzymatic assay, may be another method to determine transfection success.
In the case of siRNA, success may be measured using a reporter gene or assaying expression levels of the target mRNA (e.g., using RT-PCR) or protein (e.g., using immunoassays).
If multiple assays will be performed, make sure the techniques you choose are compatible with all assay chemistries. For example, if lysates are made from transfected cells, the lysis buffer used ideally would be compatible with all subsequent assays. In addition, if cells are needed for propagation after assessment, make sure to retain some viable cells for passage after the assay.
Factors Influencing Transfection Efficiency
With any transfection reagent or method, cell health, degree of confluency, number of passages, contamination, and DNA quality and quantity are important parameters that can greatly influence transfection efficiency. Note that with any transfection reagent or method used, some cell death will occur.
Cell Health
Cells should be grown in medium appropriate for the cell line and supplemented with serum or growth factors as needed for viability. Contaminated cells and media (e.g., contaminated with yeast or mycoplasma) should never be used for transfection. If cells have been compromised in any way, discard them and reseed from a frozen, uncontaminated stock. Make sure the medium is fresh if any components are unstable. Medium lacking necessary factors can harm cell growth. Be sure the 37°C incubator is supplied with CO2 at the correct percentage (usually 5–10%) and kept at 100% relative humidity.
If you are unsure about what type of culture medium or CO2 levels are needed for your cell line of interest, consult the American Type Culture Collection [ATCC] web site.
Confluency
As a general guideline, transfect cells at 40–80% confluency. Too few cells will cause the culture to grow poorly without cell-to-cell contact. Too many cells results in contact inhibition, making cells resistant to the uptake of foreign DNA. Actively dividing cells take up introduced DNA better than quiescent cells.
Number of Passages
Keep the number of passages low (<50). In addition, the number of passages for cells used in a variety of experiments should be consistent. Cell characteristics can change over time with immortalized cell lines, and cells may not respond to the same transfection conditions after repeated passages, resulting in poor expression of the transfected gene.
DNA Quality and Quantity
Plasmid DNA for transfections should be free of protein, RNA, chemical and microbial contamination. Suspend ethanol-precipitated DNA in sterile water or TE buffer to a final concentration of 0.2–1mg/ml. The optimal amount of DNA to use in the transfection will vary widely, depending on the type of DNA, transfection reagent, target cell line and number of cells.
Optimization of Transfection Efficiency
You will need to optimize specific transfection conditions to achieve the desired transfection efficiencies. Important parameters to consider are the charge ratio of cationic lipid transfection reagent to DNA, amount of transfected nucleic acid, length of time cells are exposed to the transfection reagent and presence or absence of serum. Reporter genes are useful to determine optimal conditions. The transfection efficiency achieved using any transfection reagents varies depending on the cell type being transfected and transfection conditions used.
Charge Ratio of Cationic Transfection Reagent to Nucleic Acid
The amount of positive charge contributed by the cationic lipid component of the transfection reagent should equal or exceed the amount of negative charge contributed by the phosphates on the DNA or RNA backbone, resulting in a net neutral or positive charge on the multilamellar vesicles associating with the DNA or RNA. Optimization of the lipid:DNA ratio is particularly important if the culture medium contains serum (Chan et al. 2014; Tranchant et al. 2004).
Nucleic Acid Amount
The optimal amount of DNA or RNA will vary depending on the type of nucleic acid, number of cells, culture dish size and target cell line used. For example, COS-7 cells are optimally transfected with 100ng of pGL4.13[luc2/SV40] Vector (Cat.# E6681) using ViaFect™ Reagent at a 4:1 ratio in a 96-well plate. In contrast, the same cells are optimally transfected with 50ng of DNA using the FuGENE® HD Transfection Reagent at a 3:1 ratio in the same well size. For other cell lines, we suggest testing the nucleic acid amounts given in Table 2.
Table 2. Suggested Nucleic Amounts to Use for Optimization.
| Transfection Reagent | Nucleic Acid Amount to Test | Reagent:Nucleic Acid Ratios to Test | Culture Dish Size |
| ViaFect™ Reagent | 0.05–0.1µg | 6:1, 5:1, 4:1, 3:1, 2.5:1 and 2:1 | 96-well plate |
| FuGENE® 6 Transfection Reagent | 0.04–0.2µg | 4:1, 3.5:1, 3:1, 2.5:1, 2:1 and 1.5:1 | 96-well plate |
| FuGENE® HD Transfection Reagent | 0.04–0.2µg | 4:1, 3.5:1, 3:1, 2.5:1, 2:1 and 1.5:1 | 96-well plate |
| FuGENE® 4K Transfection Reagent | 0.04–0.2µg | 5.5:1–3:1 | 96-well plate |
Significantly increasing the quantity of transfected nucleic may not yield better results. In fact, if initial transfection results are satisfactory, a reduced DNA or RNA quantity can be tested (while keeping the optimal reagent:nucleic ratio constant). Often a range of DNA or RNA concentrations is suitable for transfection. However, if the nucleic acid concentration is below or above this range, transfection efficiencies will decrease. If there is too little DNA or RNA, the experimental response may not be present. If there is too much nucleic, the excess can be toxic to cells. Calibrate the system using a test plasmid with reporter gene function.
Time
Traditionally, transfection reagents must remain in contact with cells for a defined period of time, after which additional medium is added or the medium is replaced to help minimize toxic effects of the reagent. The optimal transfection time depends on the cell line, transfection reagent and nucleic acid used. For the ViaFect™ Transfection Reagent and the FuGENE® HD Transfection Reagent, which are two of the more gentle methods of DNA transfection into cells, there is no need to add more medium or replace the medium after transfection.
For initial tests with liposomal reagents that require adding or replacing the medium, use a 1-hour transfection interval, and test transfection times of 30 minutes to 4 hours or even overnight, depending on the reagent used. Monitor cell morphology during the transfection interval, particularly when cells are maintained in serum-free medium, because some cell lines lose viability under these conditions.
Serum
Transfection protocols often require serum-free conditions for optimal performance because serum can interfere with many commercially available transfection reagents (Chan et al. 2014). The ViaFect™, FuGENE® HD and FuGENE® 4K Transfection Reagents can be used in transfection protocols in the presence of serum, ideal for transfecting cell types (e.g., primary cells) or applications that require continuous exposure to serum.. Note that the best results are obtained when variability is minimized among different lots of serum.
Co-Transfection and Dual-Reporter Assays
Although many experiments use a single reporter gene, a dual-reporter system has distinct advantages. A second reporter gene can normalize expression for transfection efficiency and cell number. Small perturbations in growth conditions for transfected cells can dramatically affect gene expression. A second reporter helps to determine if the effects are due to the treatment of the cells or a response from the experimental reporter (Chu and Rana 2008; Zhou et al. 2016).
The Nano-Glo® Dual-Luciferase® Reporter Assay System (NanoDLR) (Cat.# N1610) is an efficient means of quantitating luminescent signals from two reporter genes in the same sample. In this system, firefly (Photinus pyralis) and NanoLuc® luciferase activities are measured sequentially from a single sample in a homogeneous format. In the NanoDLR™ System, both reporters yield linear assay responses (with respect to the amount of enzyme) and exhibit no endogenous activity in experimental host cells. In addition, the extended half-life of the reporter signals are ideal for use with multiwell assay formats.
The various Promega Renilla luciferase vectors can be used as control vectors when co-transfected with a firefly luciferase vector into which the promoter of interest is cloned. Alternatively, the firefly vector may be used as the control vector and the NanoLuc® luciferase vector as the experimental construct. In a co-transfection experiment, note that trans effects between promoters on co-transfected plasmids can potentially affect reporter gene expression. This is primarily of concern when either the control or experimental reporter vector, or both, contain very strong promoter/enhancer elements. The occurrence and magnitude of such effects will depend on several factors: 1) the combination and activities of genetic regulatory elements present on the co-transfected vectors; 2) the amount and relative ratio of experimental vector to control vector introduced into cells; and 3) the cell type transfected.
To help ensure independent genetic expression of experimental and control reporter genes, preliminary co-transfection experiments should be performed to optimize both the vector DNA amount and ratio of co-reporter vectors. Because NanoLuc® luciferase produces an extremely bright signal, it is possible to use very small quantities of these vectors to provide low-level, constitutive co-expression of NanoLuc® luciferase activity. This means that the ratio of firefly and NanoLuc® luciferase vectors to test vector can range from 1:1 to 10,000:1 (or greater) to determine the optimal expression. The key to a dual-reporter system is to maximize expression of the experimental reporter while minimizing that of the control reporter. However, the expression level of the control reporter should be three standard deviations above background to be significant.
The strength of the promoter in your cell system is an important consideration. A more moderately expressing promoter like thymidine kinase [TK; e.g., pNL1.1.TK(Nluc/TK) ] Vector (Cat.# N1501)] may be preferable to SV40 or CMV. Stronger promoters may exhibit more trans effects, cross-talk or regulatory problems. However, adjusting the ratio of experimental vector to control vector (e.g., using 100:1 or 200:1) may eliminate some of these issues.
Controls
When transfecting nucleic acids into cells, there are controls that can help assess the transfection. These recommended controls include:
- Cells control: Medium alone (e.g., 5µl of medium)
- DNA/RNA control: DNA or RNA without transfection reagent (e.g., 100ng DNA in a 5µl volume)
- Reagent control: Transfection reagent without DNA/RNA (e.g., 0.4µl of reagent in a 5µl volume)
General Transfection Protocol
Preparing Cells for Transfection
Removing Adherent Cells Using Trypsin
Trypsinizing cells prior to subculturing or cell counting is an important technique for successful cell culture. The following technique works consistently well when passaging cells.
Materials Required:
- 1X trypsin-EDTA solution
- 1X PBS or 1X HBSS
- adherent cells to be subcultured
- appropriate growth medium (e.g., DMEM) with serum or growth factors or both added
- culture dishes, flasks or multiwell plates, as needed
- hemocytometer
- Prepare a sterile trypsin-EDTA solution in a calcium- and magnesium-free salt solution such as 1X PBS or 1X HBSS. The 1X solution can be frozen and thawed for future use, but trypsin activity will decline with each freeze-thaw cycle. The trypsin-EDTA solution may be stored for up to 1 month at 4°C.
- Remove medium from the tissue culture dish. Add enough PBS or HBSS to cover the cell monolayer: 2ml for a 150mm flask, 1ml for a 100mm plate. Rock the plates to distribute the solution evenly. Remove and repeat the wash. Remove the final wash. Add enough trypsin solution to cover the cell monolayer.
- Place plates in a 37°C incubator until cells just begin to detach (usually 1–2 minutes).
- Remove the flask from the incubator. Strike the bottom and sides of the culture vessel sharply with the palm of your hand to help dislodge the remaining adherent cells. View the cells under a microscope to check whether all cells have detached from the growth surface. If necessary, cells may be returned to the incubator for an additional 1–2 minutes.
- When all cells have detached, add medium containing serum to cells to inactivate the trypsin. Gently pipet cells to break up cell clumps. Cells may be counted using a hemocytometer, distributed to fresh plates for subculturing, or both.
Typically, cells are subcultured to prepare for transfection the next day. The subculture should bring the cells of interest to the desired confluency for transfection. As a general guideline, plate 5 × 10⁴ cells per well in a 24-well plate or 5.5 × 10⁵ cells for a 60mm culture dish for ~80% confluency on the day of transfection. Change cell numbers proportionally for different size plates (see Table 3).
Table 3. Area of Culture Plates for Cell Growth.
| Size of Plate | Growth Areaa (cm²) | Relative Areab |
| 24-well | 1.88 | 1X |
| 96-well | 0.32 | 0.2X |
| 12-well | 3.83 | 2X |
| 6-well | 9.4 | 5X |
| 35mm | 8.0 | 4.2X |
| 60mm | 21 | 11X |
| 100mm | 55 | 29X |
a This information was calculated for Corning® culture dishes.
b Relative area is expressed as a factor of the total growth area of the 24-well plate recommended for optimization studies. To determine the proper plating density, multiply 5 × 10⁴ cells by this factor.
Preparing DNA for Transfection
High-quality DNA free of nucleases, RNA and chemicals is as important for successful transfection as the transfection reagent chosen. See the DNA Purification Guide for information about purifying transfection-quality DNA.
Example Protocol Using FuGENE® 4K Reagent
We strongly recommend that you optimize transfection conditions for each cell line. If you have optimized transfection parameters, use the empirically determined conditions for your experimental transfections.
If you choose not to optimize transfection parameters, use the general conditions recommended below.
Materials Required:
- cell culture medium with serum appropriate for the cell type being transfected
- serum-free cell culture medium for complex formation (Opti-MEM® I reduced-serum medium, standard medium or sterile water can be used.)
- 96-well or other culture plates
- U- or V-bottom dilution plates or microcentrifuge tubes
- optional: 24-well culture plate to serve as a rack for FuGENE® 4K Transfection Reagent
Notes:
- The following protocol is a guideline for transfecting approximately 10–50 wells, depending on the volume of FuGENE® 4K Transfection Reagent/DNA mixture used. For additional wells, scale volume accordingly.
- The total volume of serum-free medium, DNA and FuGENE® 4K Transfection Reagent to add per well of a 96-well plate is 2–10µl. This consists of 0.04–0.2µg DNA per well, and 0.16–0.8µl FuGENE® 4K Reagent per well.
- To a sterile tube or U- or V-bottom plate, add 90–98µl of medium prewarmed to room temperature so that the final volume after adding the DNA is 100µl. Add 2µg of plasmid DNA (0.2–1µg/µl) to the medium, and mix by vortexing or pipetting. For a 4:1 FuGENE® 4K Transfection Reagent-to-DNA ratio, add 8µl of FuGENE® 4K Transfection Reagent, and mix immediately.
Notes:
a. Add FuGENE® 4K Transfection Reagent directly to medium. Do not allow undiluted FuGENE® 4K Transfection Reagent to contact the sides of the tube or U- or V-bottom plate.
b. The FuGENE® 4K Reagent-DNA complex must be prepared in medium without serum, even if the cells are transfected in the presence of serum. - Incubate the FuGENE® 4K Transfection Reagent-DNA mixture for 5–15 minutes at room temperature.
Note: We suggest starting with a 15-minute incubation. However, the optimal time may be 5–45 minutes, depending on the cell line used. Optimize the incubation time for best performance. - Add 2–10µl of the FuGENE® 4K Transfection Reagent-DNA mixture per well to a 96-well plate containing 100µl of cells in growth medium. We suggest 5µl of mixture as a starting point. Add the transfection complex to the cells in a drop-wise manner. Ensure complete mixing and even distribution over the entire surface by pipetting, using a plate shaker or rocking flasks. Return cells to the incubator for 24–72 hours.
Note: The total growth medium volume may vary, depending on well format and standard laboratory practice. - Measure transfection efficiency using an assay appropriate for the reporter gene. For transient transfection, cells are typically assayed 24–72 hours after transfection.
Optimizing Transfection with Lipid Reagents
In previous sections, we discussed factors that influence transfection success. Here we present a method to optimize transfection of a particular cell line with a single transfection reagent. For more modern lipid-based reagents such as the ViaFect™ Transfection Reagent, we recommend initially testing 50–100ng of DNA per well of a 96-well plate at reagent:DNA ratios of 3:1 or 4:1 for adherent cell lines or 2:1 for suspension cell lines. Figure 5 outlines a typical optimization matrix. When preparing the ViaFect™ Transfection Reagent:DNA complex, the incubation time may require optimization; we recommend 5–20 minutes.
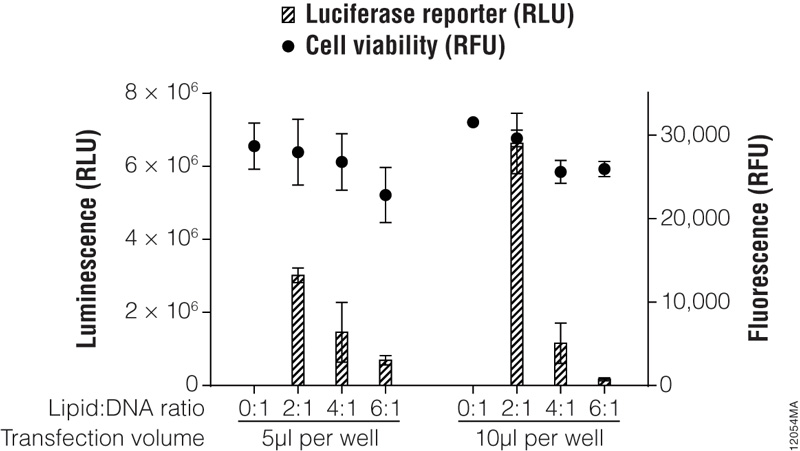
Figure 5. Transfection optimization using the ViaFect™ Transfection Reagent. TF-1 cells were plated in growth media without antibiotics at 30,000 cells per well in a white 96-well assay plate and transfected with a CMV-luc2 plasmid using various lipid (ViaFect™ Transfection Reagent):DNA ratios. The DNA concentration was held constant at 1μg per 100μl of Opti-MEM® I reduced-serum medium, and the amount of ViaFect™ Transfection Reagent was varied to obtain the indicated ratios. Either 5 or 10μl of transfection complex was then added to cells in the 96-well plate. Twenty-four hours after transfection, the ONE-Glo™ + Tox Luciferase Assay was performed. Note that 0:1 is the negative control with DNA but no lipid. These results show that, for this particular cell line, a 2:1 ViaFect™ Transfection Reagent:DNA ratio gave optimal results.
For traditional cationic lipid reagents, we recommend testing various amounts of transfected DNA (0.25, 0.5, 0.75 and 1µg per well in a 24-well plate) at two charge ratios of lipid reagent to DNA (2:1 and 4:1; see Figure 6). This brief optimization can be performed using a transfection interval of 1 hour under serum-free conditions. One 24-well culture plate per reagent is required for the brief optimization with adherent cells (three replicates per DNA amount).
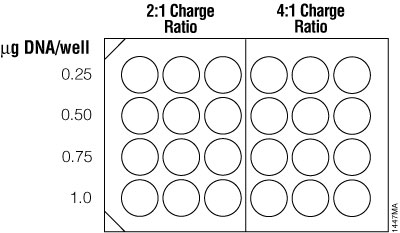
Figure 6. Suggested plating format for initial optimization of cationic lipid transfection conditions.
A more thorough optimization can be performed to screen additional charge ratios, time points and effects of serum-containing medium at the DNA amounts found to be optimal during initial optimization studies. One hour or two hours for the transfection interval is optimal for many cell lines. In some cases, however, it may be necessary to test charge ratios and transfection intervals outside of these ranges to achieve optimal gene transfer.
Some transfection methods require removing medium with reagent after incubation; others do not. Read the technical literature accompanying the selected transfection reagent to learn which method is appropriate for your system. However, if there is excessive cell death during transfection, consider decreasing time of exposure to the transfection reagent, decreasing the amounts of DNA and reagent added to cells, plating additional cells and removing the reagent after the incubation period and adding complete medium.
Endpoint Assays
Many transient expression assays use lytic reporter assays like the Dual-Luciferase® Assay System (Cat.# E1910) and Bright-Glo™ Assay System (Cat.# E2610) 24 hours after transfection. The Nano-Glo® Dual-Luciferase® Reporter Assay System (Cat.# N1610) allows detection in just a few hours after transfection. However, the time frame for most assays can vary (24–72 hours after transfection), depending on protein expression levels. Reporter-protein assays use colorimetric, radioactive or luminescent methods to measure enzyme activity present in a cell lysate. Some assays (e.g., Luciferase Assay System) require that cells are lysed in a buffer after removing the medium, then mixed with a separate assay reagent to determine luciferase activity. Others are homogeneous assays (e.g., Bright-Glo™ Assay System) that include the lysis reagent and assay reagent in the same solution and can be added directly to cells in medium. Examine the reporter assay results and determine where the greatest expression (highest reading) occurred. These are the conditions to use with your constructs of interest.
Alternative detection methods include histochemical staining of cells (determining the percentage of cells that are stained in the presence of the reporter gene substrate; Figure 7), fluorescence microscopy (Figure 8) or cell sorting if using a fluorescent reporter like the Monster Green® Fluorescent Protein phMGFP Vector (Cat.# E6421).
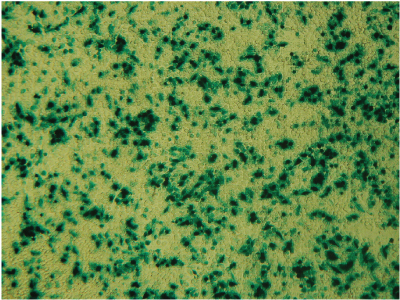
Figure 7. Histochemical staining of RAW 264.7 cells for β-galactosidase activity. RAW 264.7 cells were transfected using 0.1µg DNA per well and a 3:1 ratio of FuGENE® HD to DNA. Complexes were formed for 5 minutes prior to applying 5µl of the complex mixture to 50,000 cells/well in a 96-well plate. Twenty-four hours post-transfection, cells were stained for β-galactosidase activity using X-gal. Data courtesy of Fugent, LLC.
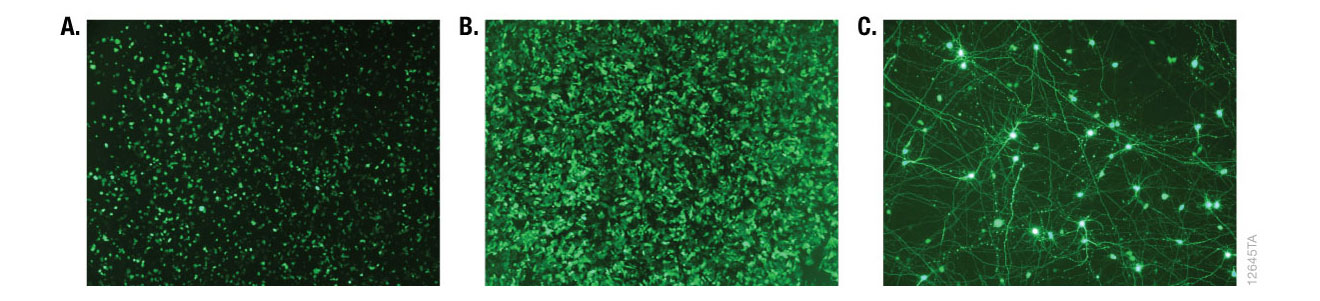
Figure 8. Fluorescence microscopy of cells transfected with ViaFect™ Transfection Reagent. iCell® human tissue cells in 96-well plates were transfected with ViaFect™ Transfection Reagent and a GFP reporter plasmid at the specified reagent:DNA ratio and GFP expression was imaged after transfection. Panel A. iCell® Hepatocytes with a 6:1 reagent:DNA ratio imaged one day after transfection. Panel B. iCell® Cardiomyocytes with a 2:1 reagent:DNA ratio imaged one day after transfection. Panel C. iCell® DopaNeurons with a 4:1 reagent:DNA ratio imaged three days after transfection. Data courtesy of Cellular Dynamics International.
Real-Time Assays
For some types of transfection experiments, especially those examining the changes in gene expression levels associated with pathological mechanisms, monitoring reporter activity in living cells is desirable. Such real-time assays can provide valuable information on the expression of multiple genes in a dynamic fashion. The Nano-Glo® Live Cell detection options (Cat.# N2011) are designed to detect NanoLuc® luminescence from living cells using nonlytic protocols. These assays monitor luminescence at a single time point or continuously for up to 72 hours without compromising cell viability.
Stable Transfection
Selecting Stably Transfected Cells
Optimization for stable transfection begins with successful transient transfection. However, cells should be transfected with a plasmid containing a gene for drug resistance, such as neomycin phosphotransferase (neo). As a negative control, transfect cells using DNA that does not contain the drug-resistance marker.
- Prior to transfection, determine the selective drug concentration required to kill nontransfected cells.
- Forty-eight hours after transfection, trypsinize adherent cells and replate at several different dilutions (e.g., 1:100, 1:500) in medium containing the appropriate selection drug. For effective selection, cells should be subconfluent since confluent, nongrowing cells are resistant to the effects of antibiotics like G-418.
- For the next 14 days, replace the drug-containing medium every 3 to 4 days.
- During the second week, monitor cells for distinct “islands” of surviving cells. Drug-resistant clones can appear in 2–5 weeks, depending on the cell type. Cell death should occur after 3–9 days in cultures transfected with the negative control plasmid.
- Transfer individual clones by standard techniques (e.g., using cloning cylinders) to 96-well plates, and continue to maintain cultures in medium containing the appropriate drug.
Table 4 provides an overview of commonly used antibiotics to select and maintain stable transfectants.
Table 4. Antibiotics Used to Select Stable Transfectants.
| Antibiotic | Resistance Gene | Working Concentration | Stock Solution |
| G-418 or Geneticin® | aminoglycoside (e.g., neomycin) phosphotransferase | G-418 is often used for initial selection at 500µg/ml with a range of 50–1,000µg/ml | 50mg/ml in either water or 100mM HEPES (pH 7.3); the latter buffer helps maintain culture medium pH |
| Hygromycin (Hygro) | hph | 10–400µg/ml | 100mg/ml in water |
| Puromycin (Puro) | pac | 1–10µg/ml | 10mg/ml in water or HEPES buffer (pH 7.0) |
Calculating Stable Transfection Efficiency
The following procedure may be used to determine the percentage of stable transfectants obtained.
Note: The stained cells will not be viable after this procedure.
Materials Required:
- methylene blue
- methanol
- cold deionized water
- light microscope
- After approximately 14 days of selection in the appropriate drug, monitor the cultures microscopically for the presence of viable cell clones. When distinct “islands” of surviving cells are visible and nontransfected cells have died out, proceed with Step 2.
- Prepare stain containing 2% methylene blue in 50–70% methanol.
- Remove the growth medium from cells by aspiration.
- Add sufficient stain to the cells to cover the bottom of the dish.
- Incubate for 5 minutes.
- Remove the stain, and rinse gently under deionized cold water. Shake off excess moisture.
- Allow the plates to air-dry. The plates can be stored at room temperature.
- Count the number of colonies, and calculate the percent of transfectants based on the cell dilution and original cell number.
Composition of Solutions
1X HBSS (Hanks Balanced Salt Solution)
|
5mM |
KCl | |
|
0.3mM |
KH2PO4 | |
|
138mM |
NaCl | |
|
4mM |
NaHCO3 | |
|
0.3mM |
Na2HPO4 | |
|
5.6mM |
D-glucose |
The final pH should be 7.1.
1X PBS
|
137mM |
NaCl | |
|
2.7mM |
KCl | |
|
4.3mM |
Na2HPO4 | |
|
1.47mM |
KH2PO4 |
The final pH should be 7.1.
1X Trypsin-EDTA solution
|
0.05% |
trypsin (w/v) | |
|
0.53mM |
EDTA |
Dissolve in a calcium- and magnesium-free salt solution such as 1X PBS or 1X HBSS.
Promega Transfection Products
FuGENE® 4K Transfection Reagent
The FuGENE® 4K Transfection Reagent (Cat.# E5911) is the most advanced, 100% synthetic, nonliposomal transfection reagent designed to transfect DNA into a variety of both challenging and routine mammalian cell lines. Provides a higher transfection efficiency than leading competitors’ reagents. This Reagent is animal-free and demonstrates a strong performance in HEK293 and CHO cells, making it ideal for protein and virus production.
Technical Bulletins and Manuals
TM694 FuGENE® 4K Transfection Reagent Technical Manual
FuGENE® HD Transfection Reagent
The FuGENE® HD Transfection Reagent (Cat.# E2311) is a novel, nonliposomal formulation to transfect DNA into a wide variety of cell lines with high efficiency and low toxicity. The protocol does not require removal of serum (including up to 100% serum) or culture medium and does not require washing or changing of medium after introducing the reagent:DNA complex. Additionally, the FuGENE® HD reagent supports transfection in chemically defined media and does not contain any animal-derived components. For more information on transfection conditions using the FuGENE® HD Transfection Reagent, visit the Transfection Assistant.
Technical Bulletins and Manuals
TM328 FuGENE® HD Transfection Reagent Technical Manual
FuGENE® 6 Transfection Reagent
The FuGENE® 6 Transfection Reagent (Cat.# E2691) is a nonliposomal reagent that transfects DNA into a wide variety of cell lines with high efficiency and low toxicity. The protocol does not require removal of serum or culture medium and does not require washing or changing of medium after introducing the reagent/DNA complex. For more information on transfection conditions using the FuGENE® 6 Transfection Reagent, visit the Transfection Assistant.
Technical Bulletins and Manuals
TM350 FuGENE® 6 Transfection Reagent Technical Manual
Promega Publications
Reverse Transfection Using FuGENE® 6 and FuGENE® HD
Optimize Transfection of Cultured Cells
ViaFect™ Transfection Reagent
ViaFect™ Transfection Reagent (Cat.# E4981) is a novel formulation designed to transfect DNA into a wide variety of cell lines with high efficiency and low toxicity. ViaFect™ Transfection Reagent has been shown to transfect cell lines that are traditionally thought of as difficult to transfect (such as hematopoietic, primary and iPSC stem cell lines) with relatively high efficiency. The protocol does not require removing serum or culture medium and does not require washing cells or changing medium after introducing the reagent/DNA complex. Additionally, ViaFect™ Transfection Reagent does not contain any animal-derived components.
Technical Bulletins and Manuals
TM409 ViaFect™ Transfection Reagent Technical Manual
Promega Publications
Alternate Protocol for Maximizing Transfection Efficiency Using ViaFect™ Reagent
Choosing the Right Transfection Reagent for Your Cell Type
TransfectNow™ HEK293 Cells
The TransfectNow™ HEK293 Cells (Cat.# NC1001) are ready-to-use cells that offer several advantages over maintaining cells in continuous culture. By simply thawing, diluting, and transfecting, one can save time needed to both expand cells and maintain cell-line integrity.
Technical Bulletins and Manuals
TM690 TransfectNow™ HEK293 Cells Technical Manual
References
- An, H. and Jin, B. (2012) Prospects of nanoparticle-DNA binding and its implications in medical biotechnology. Biotechnol. Adv. 30, 1721–32.
- Bockamp, E. et al. (2002) Of mice and models: Improved animal models for biomedical research. Physiol. Genomics 11, 115–32.
- Boussif, O. et al. (1995) A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 92, 7297–301.
- Burkholder, J.K. et al. (1993) Rapid transgene expression in lymphocyte and macrophage primary cultures after particle bombardment-mediated gene transfer. J. Immunol. Meth. 165, 149–56.
- Cappechi, M.R. (1980) High efficiency transformation by direct microinjection of DNA into cultured mammalian cells. Cell 22, 479–88.
- Chan, C.-L. et al. (2014) Optimizing cationic and neutral lipids for efficient gene delivery at high serum content. J. Gene Med. 16, 84–96.
- Chu, C.-Y. and Rana, T.M. (2008) Potent RNAi by short RNA triggers. RNA 14, 1714–9.
- Chuang, C.-K. et al. (2017) Generation of mutant pigs by direct pronuclear microinjection of CRISPR/Cas9 plasmid vectors. Bio-Protocol 7, e2321.
- Cullis, P.R. and Hope, M.J. (2017) Lipid nanoparticle systems for enabling gene therapies. Mol. Ther. 25, 1467–75.
- Ding, X. et al. (2017) High-throughput nuclear delivery and rapid Expression of DNA via mechanical and electrical cell-membrane disruption. Nat. Biomed. Eng. 1, 0039.
- Dziegiel, N. (2016) Nanoparticles as a tool for transfection and transgenesis—a review. Ann. Anim. Sci. 16, 53–64.
- Farhood, H. et al. (1995) The role of dioleoyl phosphatidylethanolamine in cationic liposome mediated gene transfer. Biochim. Biophys. Acta 1235, 289–95.
- Felgner, P.L. et al. (1987) Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 84, 7413–7.
- Felgner, P.L. (1990) Particulate systems and polymers for in vitro and in vivo delivery of polynucleotides. Adv. Drug Deliv. Rev. 5, 163–87.
- Felgner, P.L. et al. (1995) Improved cationic lipid formulations for in vivo gene therapy. Ann. NY Acad. Sci. 772, 126–39.
- Fraley, R. et al. (1980) Introduction of liposome-encapsulated SV40 DNA into cells. J. Biol. Chem. 255, 10431–5.
- Gao, X. and Huang, L. (1995) Cationic liposome-mediated gene transfer. Gene Ther. 2, 710–22.
- Graham, F.L. and van der Eb, A.J. (1973) A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 52, 456–67.
- Groskreutz, D. and Schenborn, E.T. (1997) Reporter systems. In: Methods in Molecular Biology 63, 11ed. R. Tuan, Humana Press, NJ.
- Kabanov, A.V. and Kabanov, V.A. (1995) DNA complexes with polycations for the delivery of genetic material into cells. Bioconjugate Chem. 6, 7–20.
- Kawai, S. and Nishizawa, M. (1984) New procedure for DNA transfection with polycation and dimethyl sulfoxide. Mol. Cell. Biol. 4, 1172–4.
- Kim, T.K. and Eberwine, J.H. (2010) Mammalian cell transfection: the present and the future. Anal. Bioanal. Chem. 397, 3173–8.
- Klein, T.M. et al. (1987) High-velocity microprojectiles for delivering nucleic acids into living cells. Nature 327, 70–3.
- Kukowska-Latallo, J.F. et al. (1996) Efficient transfer of genetic material into mammalian cells using Starburst polyamidoamine dendrimers. Proc. Natl. Acad. Sci. USA 93, 4897–902.
- Labat-Moleur, F. et al. (1996) An electron microscopy study into the mechanism of gene transfer with lipopolyamines. Gene Ther. 3, 1010–7.
- McCutchan, J.H. and Pagano, J.S. (1968) Enhancement of the infectivity of simian virus 40 deoxyribonucleic acid with diethylaminoethyl-dextran. J. Natl. Cancer Inst. 41, 351–7.
- Ogura, R. et al. (2005) Multi-color luciferases as reporters for monitoring transient gene expression in higher plants. Plant Biotechnol. 22, 151–5.
- Sarver, N.P. et al. (1981) Bovine papilloma virus deoxyribonucleic acid: A novel eucaryotic cloning vector. Mol. Cell. Biol. 1, 486–96.
- Schimke, R.T. (1988) Gene amplification in cultured cells. J. Biol. Chem. 263, 5989–92.
- Sessa, G. and Weissmann, G. (1968) Phospholipid spherules (liposomes) as a model for biological membranes. J. Lipid Res. 9, 310–8.
- Shigekawa, K. and Dower, W.J. (1988) Electroporation of eukaryotes and prokaryotes: A general approach to the introduction of macromolecules into cells. BioTechniques 6, 742–51.
- Stewart, M.P. et al. (2016) In vitro and ex vivo strategies for intracellular delivery. Nature 538, 183–92.
- Tranchant, I. et al. (2004) Physicochemical optimisation of plasmid delivery by cationic lipids. J. Gene Med. 6, S24–S35.
- Vaheri, A. and Pagano, J.S. (1965) Infectious poliovirus RNA: A sensitive method of assay. Virology 27, 434–6.
- Wall R.J. (2001) Pronuclear microinjection. Cloning Stem Cells 3, 209–20.
- Wong, T.K. and Neumann, E. (1982) Electric field mediated gene transfer. Biochem. Biophys. Res. Commun. 107, 584–7.
- Ye, G.N. et al. (1990) Optimization of delivery of foreign DNA into higher-plant chloroplasts. Plant Mol. Biol. 15, 809–19.
- Zhou, X. et al. (2016) MicroRNA-761 is upregulated in hepatocellular carcinoma and regulates tumorigenesis by targeting Mitofusin-2. Cancer Sci. 107, 424–32.