High-Quality RNA from Peripheral Blood and Bone Marrow Samples After Automated Extraction on a Maxwell®16 Instrument
The Maxwell® 16 LEV simplyRNA Blood Kit was used for isolation of total RNA from whole blood or bone marrow samples. The kit is designed to extract high-quality RNA that can be used in many downstream RNA applications. This procedure involves minimal sample handling before the automated purification on the Maxwell® 16 Instrument.
Ludovica Riera1,2, Francesca Sismondi2, Barbara Nuschak2, Maria Bonavita1,2, Maria Antonietta De Felice2, Mariano Pace2, Giorgio Inghirami1,2, Paola Francia di Celle2.
1Department of Molecular Biotechnology and Health Sciences, Section of Pathology, University of Turin, Italy; 2Center for Experimental Research and Medical Studies (CeRMS), AO Città della Salute e della Scienza, Turin, Italy.
Publication Date: October 2013; tpub 126
Abstract
RNA purification is one of the most critical steps in assays used to monitor changing expression (both qualitative and quantitative) within blood cells. Isolation of RNA is complicated by the fact that RNA is significantly more labile than DNA, making RNA more challenging to purify. Moreover, the use of low-quality RNA may compromise the results of downstream applications, which can cause expensive and time-consuming assay repeats.
Here we describe a method to improve the quality and reproducibility of RNA samples and to identify an automated method that is simple and risk-free for operators, using the Maxwell® 16 LEV simplyRNA Blood Kit for RNA purification from peripheral blood or bone marrow samples on the automated Maxwell® 16 Instrument.
Introduction
Maintenance of RNA integrity throughout preparation is essential in obtaining meaningful gene expression data in molecular biology laboratories. RNA integrity is particularly important for quantitative assays where evaluation of an internal control gene is essential for assessing and monitoring sample quality. The suitability of RNA samples is strongly dependent on the source of the RNA.
RNA purification is one of the most critical steps in assays used to monitor changing expression (both qualitative and quantitative) within blood cells. Isolation of RNA is complicated by the fact that RNA is significantly more labile than DNA, making RNA more challenging to purify. Moreover, the use of low-quality RNA may compromise the results of downstream applications, which can cause expensive and time-consuming assay repeats.
Recently, there has been an increased effort to standardize molecular monitoring procedures and guidelines for analysis of results. Nevertheless, many available and reliable RNA purification methods are difficult to implement and labor-intensive due to the complexity of the manual isolation procedures.
Here we describe a method to improve the quality and the reproducibility of RNA samples and to identify an automated method that is simple and risk-free for operators, using the Maxwell® 16 LEV simplyRNA Kit (Cat.# AS1270) for RNA purification from peripheral blood or bone marrow samples on the automated Maxwell® 16 Instrument.
Successful use of the Maxwell® 16 Instrument in RNA purification was determined by successful amplification in two separate real-time PCR (qPCR) assays and analysis of the RNA quality on a 2100 Bioanalyzer Instrument (Agilent Technologies Cat.# G2938C, G2946CA).
Methods
RNA was isolated from a total of 450 samples, 245 peripheral blood (PB) samples and 205 bone marrow (BM) samples.
Isolation of Bone Marrow Mononuclear Cells
Separation of lymphocytes and monocytes from other blood components was performed using cell separation media (Lympholyte, Cedarlane Cat.# CL5020).
- Transfer 3ml of blood with sodium citrate as anticoagulant into a sterile 15ml tube.
- Dilute the blood with sterile saline solution to 10ml.
- Add 5ml of cell separation media to a 15ml centrifuge tube. Carefully pour the diluted blood onto the cell separation media solution.
- Centrifuge the tubes for 20 minutes at 2,000 × g (Heraeus Megafuge 1.0R).
- Harvest the layer containing the white blood cells.
- Dilute and wash the white blood cells twice with 10ml of saline solution.
- Transfer the lymphomonocyte pellet to cryotubes. Add 200μl of chilled 1-Thioglycerol/Homogenization Solution (included in the Maxwell® 16 LEV simplyRNA Blood Kit). Mix well. Freeze the resuspended cells at
–80°C if samples are not processed immediately.
Isolation of Total White Blood Cells from Whole Peripheral Blood
- Transfer 2.5ml of fresh whole blood from an EDTA collection tube into a sterile 15ml tube.
- Add 7.5ml of Cell Lysis Solution and invert the tube 5–6 times to mix. The red blood cells are lysed, leaving the white blood cells intact.
- Incubate lysates for 10 minutes at room temperature. Mix twice during the incubation by inverting the tube.
- Centrifuge tubes at 3,000 × g for 10 minutes (Heraeus Megafuge 1.0R).
- Discard as much of the supernatant as possible without disturbing the visible white pellet.
- Add 200μl of chilled 1-Thioglycerol/Homogenization Solution to the pellet. Mix well with a pipette.
- Freeze and store pellets at –80°C if samples are not immediately processed.
RNA Extraction and Purification
- Thaw samples that were frozen.
- Add 200μl of Lysis Buffer (MC501C, included in the Maxwell® 16 LEV simplyRNA Blood Kit) and 25μl of Proteinase K (Cat.# V3021) to the suspended pellet. Mix by vortexing for 20 seconds.
- Incubate at room temperature for 10 minutes.
- Add 10μl of DNase I solution (Z358A, included in the Maxwell® 16 LEV simplyRNA Blood Kit) to well #4 of the simplyRNA Blood cartridge.
- Add lysate (425µl) to well #1 of the simplyRNA Blood cartridge.
- Run the simplyRNA Blood protocol on the Maxwell® 16 Instrument.
- At the end of protocol, RNA was eluted in 40μl of Nuclease-Free Water to the 0.5ml elution tubes in the front of the Maxwell® 16 LEV cartridge rack.
An additional particle capture step was performed using the 0.5ml MagneSphere® Technology Magnetic Separation Stand (Cat.# Z5331) to remove any residual paramagnetic particles. For samples that are not processed immediately, the eluted RNA can be stored at –80°C.
Results
Purified RNA samples were quantitated by absorbance using a NanoDrop® ND-1000 instrument (Thermo Scientific Cat.# NDR ND1000, NDB494). The average RNA yield for PB was 216ng/µl and for BM was 415ng/µl in a 40μl volume. Purity was determined by measuring absorbance at 260 and 280nm and calculating the A260/280 ratio.
RNA Integrity
A bioanalyzer (Agilent 2100 Bioanalyzer Instrument) was used to determine RNA integrity numbers (RIN) as a measure of RNA quality, with numbers >8 representing good-quality RNA (1) . Representative PB samples (n=23) were analyzed using the Agilent RNA 6000 Nano Kit (Agilent Technologies, Cat.# G2938-90035). Samples had mean RIN values >9.3 with a range of 9–9.8, indicating high-quality RNA. Examples of electrophoresis profile and RNA quality parameters are shown in Figure 1, Panels A and B.
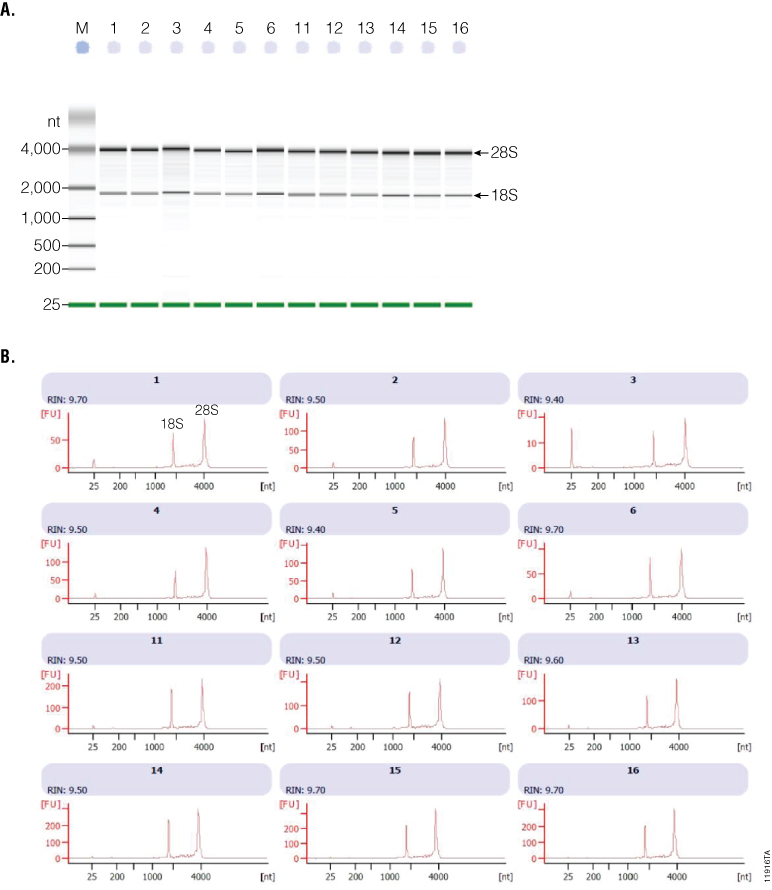
Figure 1. Agilent 2100 bioanalyzer electrophoresis (Panel A) and analysis (Panel B) file run summary detailing the RNA quality features in 16 representative samples. Panel A. The bioanalyzer provides a better assessment of RNA integrity (subunits 18S and 28S) by showing a detailed picture of the size distribution of RNA fragments. Panel B. The peaks corresponding to the ribosomal 18S and 28S RNA are part of the algorithm for the RIN calculation used as a measure of the RNA quality (RIN; 1 = totally degraded, 10 = intact).
cDNA Synthesis
Total RNA extracted from cells was used as a template for cDNA transcription using SuperScript® III First-Strand Synthesis SuperMix for RT-qPCR (Life Technologies Corporation Cat.# 18080-400) (2), (3). The protocol formulation can be used to quantify fewer than 10 copies of a target gene using RT-qPCR, and has a broad dynamic range that supports accurate quantification of high-copy mRNA in up to 1μg of total RNA.
Master mix for cDNA synthesis:
- 2X RT Reaction Mix 10μl
- RT Enzyme Mix 2μl
- RNA (up to 1 μg) xμl
- DEPC-treated water to 20μl
Treatment with E. coli RNase H in a final volume of 50μl was used to remove the RNA template from the cDNA:RNA hybrid molecule after first-strand synthesis.
Quantitative Real-Time PCR (qPCR) Protocols and Results
To determine if the purified RNA samples could be efficiently reverse-transcribed and amplified, the samples were assayed using quantitative PCR methods without post-PCR processing. We used real-time detection of fluorescent signals during PCR cycling, significantly reducing the risk of PCR product contamination (4) , (5) . Two types of real-time qPCR instruments, iCycler IQ® Real-Time PCR Detection System (Bio-Rad Cat.# 170-8740) and Applied Biosystems 7300 Real-Time PCR instrument (LifeTechnologies, Cat.# SC7300) were used to evaluate ABL control gene copy number in each sample.
Samples (n=450; 205 BM and 245 PB) were screened on the iCycler IQ® apparatus and ABL real-time qPCR assays were performed using 2X IQ® Supermix (Bio-Rad Cat.# 170-8862) and ABL standards (Qiagen Cat.# 674691).
In addition, some samples (n=251; 138 BM and 113 PB) underwent real-time qPCR analysis for ABL detection on Applied Biosystems platform using ABL mix from ProfileQuant® kit (Qiagen Cat.# 676923) or from Alert kit (ELITechGroup, Cat.# RTSG07-210).
The standard curve was derived from three dilution points, 105–103 for Qiagen standards, as values below 103 are not acceptable for ABL, while a 102 (as in four dilution points for ELITechGroup ABL standards) dilution may be of limited use for ABL. The threshold is used to determine the threshold cycle (Cq) and is typically set in the log-linear phase of the amplification curve, where the Cq value denotes the PCR cycle at which fluorescence is detected above the background level.
Each qPCR run should conform to the following criteria:
- Slope –3.2 to –3.6
- R2 > 0.980
Runs that do not meet these criteria should be rejected.
Amplification of an endogenous control gene such as ABL is used as a reference gene to normalize target transcript levels and identify poor quality samples. ABL is the most common reference gene used due to its wide expression level across different cell types and its stability. The ability to amplify at least 10,000 ABL molecules per reaction provides, in our experience, a safety measure against poor sample quality, avoiding incorrect normalization in quantitative assay and also false-negative results. All data obtained were included in the analysis. By assessing the absolute value of the ABL housekeeping gene we estimated whether quantification results could be affected by this purification method.
qPCR results on both platforms are summarized in Figure 2 by reporting the mean ABL copy number. As shown, the housekeeping gene was detected with a value ranging from 2.59 x 104 (column 6) and 5.79 x 104 (column 3). Significant differences between the two methods were detected on peripheral blood only but still maintaining mean ABL copy number above 10,000, indicating high quality of the RNA. As shown in Figure 2, a slight difference in values obtained with the two RQ-PCR apparatus is always detected, probably due to the use of different reagents and protocols on the two instruments.
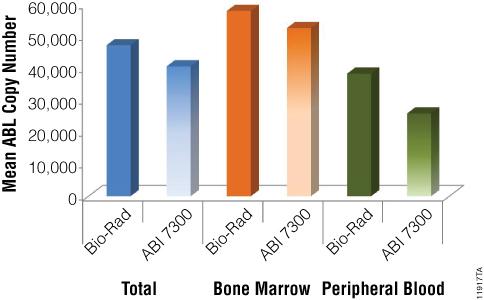
Figure 2. Mean ABL copy number in all samples analyzed with two different qPCR systems (Bio-Rad and ABI 7300).
Indeed, as shown in Figure 3, all samples revealed good RNA quality, with excellent performance in qPCR and nearly all of the samples (above 95%) showed an ABL copy number above 10,000. In less than 5% of samples ABL copy number was between 1,000 and 10,000 copies, thus resulting samples are suitable for most qualitative downstream applications.
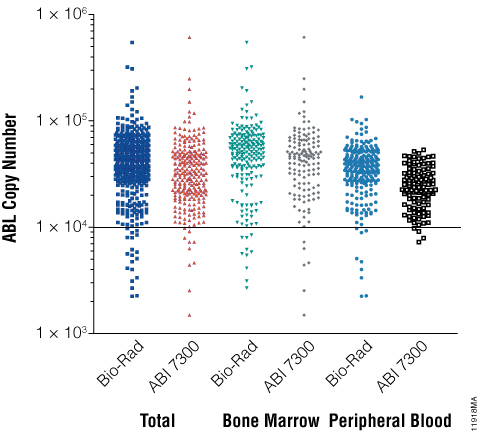
Figure 3. Sample quality distribution measured by ABL copy number with two different qPCR systems (Bio-Rad and ABI 7300). ABL copy number was higher than 10,000 in >95% of samples.
The improvements obtained by the use of an automated apparatus could be affected by possible contamination between samples due to purification cartridges with open configuration. Therefore, we analyzed samples simultaneously extracted in a Maxwell® purification session; some of them were positive for a fusion transcript and the results corresponded to the expected data. We did not find cross-contamination in this nested PCR assay (data not shown), demonstrating the safety of the prefilled purification cartridges placed in the instrument.
Discussion
In this study we isolated RNA using either 2.5ml of peripheral blood or cells from bone marrow after cell pellets were prepared using gradient cell separation media. We modified the protocol to use a smaller elution volume in order to obtain more concentrated RNA. RNA quality was evaluated by absorbance using the A260/280 ratio, bioanalyzer readings and qPCR analysis.
Here, the automated Maxwell® 16 LEV simplyRNA Kit (Cat.# AS1270) gave RNA with high yield and quality that fulfilled the qPCR requirements. In addition, the bioanalyzer analysis revealed optimal RNA integrity in all tested samples.
We did not see many red blood cells in the pellet nor foaming of the samples (neither PB nor BM) during vortex mixing. At times the cell pellet in Homogenization Solution was too viscous, depending on the number of cells in the sample. As suggested by extraction kit protocol, you can reduce this viscosity by adding an additional 100μl of Homogenization Solution with 1-Thioglycerol. Moreover, we found neither RNA degradation nor low RNA yield to be a problem in terms of downstream reverse transcription processing. Finally, we found no cross-contamination between samples, which indicates the extraction method is suitable for preparing RNA for analysis in downstream molecular applications.
One limitation of this system is the restricted number of samples (1–16) that can be simultaneously processed on the Maxwell® 16 Instrument. However, this limitation can be overcome by performing repeated extraction sessions in the same day.
All samples extracted, from both bone marrow and peripheral blood, were suitable for qualitative and mutational molecular assays, and more than 95% of the samples revealed RNA quality suitable for quantitative analysis.
The possibility of using an automated system for sample preparation like the one we have just successfully described may represent a key point for standardization of extraction procedures in order to achieve increasingly reproducible, sensitive and reliable results.
References
- Schroeder, A. et al. (2006) The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Molecular Biology 7, 3.
- Kotewicz, M.L., D'Alessio, J.M., Driftmier, K.M., Blodgett, K.P., and Gerard, G.F. (1985) Cloning and overexpression of Moloney murine leukemia virus reverse transcriptase in Escherichia coli. Gene 35 , 249.
- Gerard, G.F., D'Alessio, J.M., Kotewicz, M.L., and Noon, M.C. (1986) Influence on stability in Escherichia coli of the carboxy-terminal structure of cloned Moloney murine leukemia virus reverse transcriptase. DNA 5 , 271–9.
- Wong, M.L, Medrano, J.F. (2005) Real-time PCR for mRNA quantitation. Biotechniques 39, 75-85.
- Huggett, J., Dheda, K., Bustin, S., Zumla, A. (2005) Real-time RT-PCR normalisation; strategies and considerations. Genes Immun 6, 279-84.