Long PCR Amplification of p300 cDNA Using a Blend of Taq DNA Polymerase and Proofreading Pfu DNA Polymerase
MCB1, Leiden University Medical Center, Wassenaarseweg 72, Leiden, The Netherlands
Publication Date: 2000
Abstract
Amplification of a large p300 cDNA was accomplished using a combination of Taq DNA Polymerase and the proofreading Pfu DNA Polymerase (Cat.# M7741). The resulting 7.5kb DNA fragment was resolved on an agarose gel.
Introduction
Transformation of cells by human adenoviruses is accomplished by alterations in cellular gene expression. Essential for the transformation is the adenovirus-E1A protein. The E1A protein exerts its effect by binding to cellular proteins, like the retinoblastoma protein and the p300 protein. The p300 protein is known to be a transcriptional coactivator for various transcription factors (1) . In our studies we focus on clarifying the role of the binding of E1A to p300 in the transformation process. For this study, E1A and p300 expression constructs were needed. We used PCR amplification as the first step in making these expression constructs, as described for the 7.5kb p300 cDNA in this report.
It is generally known that amplification of DNA fragments larger than 1kb with Taq DNA polymerase is problematic. Lack of proofreading capacity allows misincorporations, which reduce the efficiency of amplification. Amplification with a proofreading thermostable DNA polymerase removes misincorporated nucleotides (2) . However, the yield of amplification product typically drops with increasing DNA length.
The combination of Taq DNA polymerase and a low level of thermostable DNA polymerase that exhibits a 3´-exonuclease activity (proofreading DNA polymerase, e.g., Pfu DNA polymerase) has been shown to successfully amplify DNA fragments up to 35kb (2) . This PCR amplification of large DNA templates results in specific amplification products in sufficient amounts for downstream applications. This approach has been applied to the 7.5kb p300 cDNA using a blend of Taq/Pfu DNA Polymerase.
Long PCR Amplification
A DNA polymerase mix was made by combining 5 units of Taq DNA Polymerase and 0.2 units of Pfu DNA Polymerase (Cat.# M7741). The amplification reaction consisted of 1X Pfu DNA Polymerase Reaction Buffer, 250µM of each dNTP, 1µM each of sense and antisense oligonucleotide primers, 50ng of plasmid containing p300 cDNA and 1µl of the DNA polymerase mix in a total reaction volume of 50µl.
The amplification profile consisted of an initial denaturation at 94°C for 6 minutes followed by 25 cycles of denaturation (94°C for 1 minute), annealing (50°C for 1 minute) and extension (72°C for 16 minutes, approximately 2 minutes for every kilobase pair amplified). A final extension of 8 minutes at 72°C was performed.
After amplification 10µl of the reaction mixture was tested on a 1% agarose gel containing ethidium bromide. Amplified fragments were visualized by UV light. Fragment size was estimated by inclusion of lambda HindIII/EcoRI markers.
Results
Figure 1 shows the results of the amplification. A fragment of the predicted size (7.5kb) could be seen; no other fragments were observed.
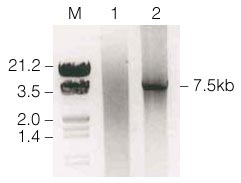 Figure 1. p300 PCR product following amplification with Taq and Pfu DNA Polymerases.
Figure 1. p300 PCR product following amplification with Taq and Pfu DNA Polymerases.
Lane M, 3µg lambda Hind III/EcoRI markers; lane 1, 10µl of PCR product from the Taq/Pfu DNA Polymerase) blend with degraded p300 cDNA clone; lane 2, 10µl of PCR product from the Taq/Pfu DNA Polymerase blend with intact p300 cDNA clone.
Discussion
Previously, PCR amplification of total p300 cDNA with Taq DNA polymerase was hampered by the large size of the cDNA. Generally low or no yield is obtained and mutations are introduced. However, the ability to PCR amplify long fragments with high specificity would speed and simplify the analysis of the complete p300 cDNA. A plasmid containing p300 cDNA was used as a template in a PCR amplification. Total length of this plasmid was 13kb. Using the protocol described in this report, a 7.5kb fragment could be amplified from this plasmid; this is the expected length of the p300 cDNA. Amplification of the 7.5kb fragment with no apparent nonspecific amplification products suggests that the procedure was specific for the target DNA.
Here we show the usefulness of a blend of Taq DNA Polymerase and the proofreading Pfu DNA Polymerase to obtain long DNA templates with high specificity. In this study a blend of 5 units of Taq DNA Polymerase and 0.2 units of Pfu DNA Polymerase resulted in optimal amplification results.
 Watch this animated, step-by-step introduction to PCR--a landmark molecular biology technique.
Watch this animated, step-by-step introduction to PCR--a landmark molecular biology technique.
Related Resources
Article References
- Eckner, R. et al. (1994) Molecular cloning and functional analysis of the adenovirus E1A-associated 300-KD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 8, 869–84.
- Cheng, S. et al. (1994) Effective amplification of long targets from cloned inserts and human genomic DNA. Proc. Natl. Acad. Sci. USA 91, 5695–9.
How to Cite This Article
Scientific Style and Format, 7th edition, 2006
Martens, J. Long PCR Amplification of p300 cDNA Using a Blend of Taq DNA Polymerase and Proofreading Pfu DNA Polymerase. [Internet] 2000. [cited: year, month, date]. Available from: https://www.promega.com/resources/pubhub/enotes/long-pcr-amplification-of-p300-cdna-using-a-blend-of-taq-and-proofreading-pfu-dna-polymerase/
American Medical Association, Manual of Style, 10th edition, 2007
Martens, J. Long PCR Amplification of p300 cDNA Using a Blend of Taq DNA Polymerase and Proofreading Pfu DNA Polymerase. Promega Corporation Web site. https://www.promega.com/resources/pubhub/enotes/long-pcr-amplification-of-p300-cdna-using-a-blend-of-taq-and-proofreading-pfu-dna-polymerase/ Updated 2000. Accessed Month Day, Year.
Products may be covered by pending or issued patents or may have certain limitations. Please visit our Web site for more information.