Epigenetics Overview
For ordering information on the products discussed here, please visit our Epigenetics Research product pages.
Introduction to Epigenetics
Epigenetics refers to heritable changes in gene expression that arise from changes in chromosomes without alteration of DNA sequence. These changes occur throughout all stages of development or in response to environmental factors such as exposure to toxins or chronic stress and are implicated in diseases such as cancer. Epigenetic mechanisms of gene regulation, which collectively make up the epigenome, include modifications to DNA and histone components of nucleosomes as well as expression of noncoding RNAs (ncRNAs). These modifications can affect gene accessibility to DNA-binding and regulatory proteins such as methyl-CpG-binding proteins, transcription factors, RNA polymerase II and other components of the transcriptional machinery, ultimately altering transcription patterns, often in tissue- and cell-specific ways. A schematic diagram showing the most well characterized epigenetic modifications are shown in Figure 1.
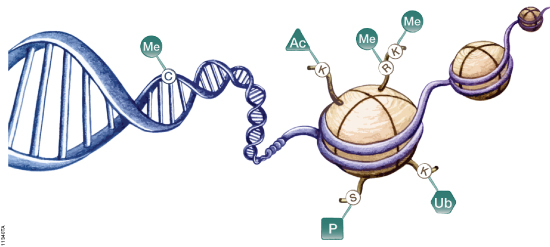
Figure 1. Epigenetic mechanisms involved in regulation of gene expression. Cytosine residues within DNA can be methylated, and lysine and arginine residues of histone proteins can be modified. Me = methylation, Ac = acetylation, P = phosphorylation, Ub = ubiquitination.
DNA Methylation
In vertebrates, DNA methylation occurs on the 5C position of cytosine residues to yield 5-methylcytidine. This occurs almost exclusively within CpG dinucleotides, although nonCpG methylation does occur in plants (primarily CpNpG and CpHpH methylation, where H = A,T,C) and to a lesser extent, mammals. Other forms of cytosine exist, including 5-hydroxymethylcytosine, 5-formylcytosine and 5-carboxylcytosine (Kriaucionis and Heintz, 2009; Ito et al. 2011; Pfaffeneder et al. 2011), which may be intermediates in a pathway for DNA demethylation.
In mammalian genomes, approximately 70–80% of CpG dinucleotides are methylated. However, stretches of CpG-rich sequences with low levels of DNA methylation, known as CpG islands, exist (reviewed in Blackledge and Klose, 2011; Deaton and Bird, 2011). DNA methylation is typically associated with epigenetic gene repression, and many targets of de novo DNA methylation during differentiation are promoters of stem cell- and germline-specific genes (Weber et al. 2007; Mohn et al. 2008; Farthing et al. 2008). DNA methylation also recruits methyl-CpG-binding proteins, which recruit additional proteins that add silencing modifications to neighboring histones. This coordination between DNA methylation and silencing histone marks leads to compaction of chromatin and gene repression.
CpG Islands
CpG islands (CGIs) make up only 0.7% of the human genome but contain 7% of the CpG dinucleotides. CpG islands often are highly enriched at gene promoters, and approximately 60% of all mammalian gene promoters are CpG-rich. CpG islands are typically unmethylated, open regions of DNA with low nucleosome occupancy. As such, CpG islands promote relaxed chromatin structure that favors active transcription, known as euchromatin, and increases accessibility of RNA polymerase II and other components of the basal transcription machinery to the transcription start site. Most CGI promoters have heterogeneous transcription start sites and lack TATA boxes, so transcription factors with CpG in their recognition sites, such as SP1, can help recruit TATA-binding protein to promoters without TATA boxes. Without additional regulatory signals, transcription from CGI promoters results in nonproductive, bidirectional cycles of initiation and premature termination. The regulatory signals required for the transition from this nonproductive state to productive, directional synthesis of full-length transcripts are not yet well characterized.
The mechanisms that keep CpG islands free of methylation appear to involve binding of transcription factors and other transcriptional machinery or the act of transcription itself. However, CpG islands can become hypermethylated (Meissner et al. 2008; Mohn et al. 2008) to silence specific genes during cellular differentiation, genomic imprinting and X chromosome inactivation.
DNA Methylases
DNA methylation is catalyzed by DNA methyltransferases (DNMTs; reviewed in Carey et al. 2011). DNMT3A and DNMT3B are involved in de novo methylation (Okano et al. 1999) and are targeted to particular genomic regions by specific histone modifications. During DNA replication, the protein Np95 recognizes hemimethylated DNA and directs DNMT1 to the replication fork to maintain patterns of DNA methylation (Pradhan et al. 1999).
Techniques to Assess DNA Methylation
Methylation-Sensitive Restriction Enzymes
The methylation status of a DNA sequence can be determined using a variety of techniques such as the use of restriction enzymes (REs), which recognize short DNA sequences and cleave double-stranded DNA at specific sites within or adjacent to these sequences. Some REs are sensitive to methylation and will not cleave DNA if a cytosine in their recognition sites is methylated, while other REs are insensitive to methylation. The methylation-sensitive RE HpaII was used in early epigenetics studies to determine that 55–70% of all HpaII sites (5'-CCGG-3') are methylated in the mammalian genome (Bird, 1980; Bestor et al. 1984) and to identify CpG-rich, hypomethylated DNA regions [known as HpaII tiny fragments (HTFs); Bird, 1986].
A list of REs that are sensitive to CpG and CpNpGp methylation can be found in the Technical Reference section of the Promega web site
Pairs of isoschizomers where one RE is insensitive to methylation and the other is sensitive (Table 1) are often used to query methylation status. DNA fragments generated by a methylation-sensitive isoschizomer will differ in size from fragments generated by a methylation-insensitive isoschizomer. The extent of cytosine methylation can be estimated by calculating the ratio of the different DNA fragments.
| Table 1. Methylation Sensitivity of Isoschizomer and Neoschizomer Pairs. | ||
| Methylated Sequence | Cleaved by | Not Cleaved by |
| m4CCGG | MspI (C/CGG) | HpaII (C/CGG) |
| Cm5CGG | MspI (C/CGG) | HpaII (C/CGG) |
| Cm4CGG | MspI (C/CGG) | HpaII (C/CGG) |
| CCm5CGGG | XmaI (C/CCGGG) | SmaI (CCC/GGG) |
| Gm6ATC | Sau3AI (/GATC) | MboI, NdeII (/GATC) |
| GATm5C | MboI, NdeII (/GATC) | Sau3AI (/GATC) |
| GATm4C | MboI (/GATC) | Sau3AI (/GATC) |
| GGTACm5C | KpnI (GGTAC/C) | Acc65I (G/GTACC) |
For more information about restriction enzymes, visit the Promega Restriction Enzyme Guide.
Bisulfite Sequencing
Bisulfite sequencing refers to techniques that assess DNA methylation through bisulfite conversion, which converts unmethylated cytosine residues to uracil residues. Methylated cytosine residues remain unmodified (Frommer et al. 1992). The target DNA is purified, alkaline- or heat-denatured, treated with sodium bisulfite, cleaned up, treated with alkaline, then cleaned up again to remove salts and other components that can inhibit downstream applications. DNA purification kits, such as the Wizard® SV Gel and PCR Clean-Up System (Cat.# A9281), are commonly used for this purpose. After bisulfite conversion and DNA cleanup, the DNA is amplified by whole genome PCR, and the amplified products are analyzed using a technique that distinguishes products derived from unmethyled DNA, which contain thymine residues, from products derived from methylated DNA, which contain cytosine residues. These techniques include pyrosequencing, methylation-specific PCR, methylation-sensitive single-strand conformation analysis (MS-SSCA; Bianco et al. 1999), high-resolution melting analysis (Wojdacz and Dobrovic, 2007), methyl cytosine immunoprecipitation (mCIP; Zhang et al. 2006), bisulfite methylation profiling (BiMP; Reinders et al. 2008) and MALDI-TOF mass spectrometry (Schatz et al. 2006). For high-throughput analysis, bisulfite-treated DNA can be analyzed using microarrays with two sets of oligonucleotide probes, one of which is complementary to cytosine-containing DNA and the other complementary to thymine-containing DNA.
Typical bisulfite conversion protocols involve long incubation times under harsh conditions, resulting in highly fragmented DNA. Promega offers the MethylEdge™ Bisulfite Conversion System (Cat.# N1301), which results in efficient DNA conversion and recovery with reduced template fragmentation using a protocol that can be completed in less than two hours, including desulphonation and cleanup. The MethylEdge™ Bisulfite Conversion System does not require an additional cleanup kit.
Additional Resources for Bisulfite Conversion
Technical Bulletins and Manuals
TM381
MethylEdge™ Bisulfite Conversion System Technical
Manual
TB308
Wizard® SV Gel and PCR Clean-Up
System Technical Bulletin
Luciferase-Based Sensors of DNA Methylation
The firefly luciferase reporter protein (Fluc) can be used to assess DNA methylation at the genome level or at specific DNA sequences. Researchers have developed split-luciferase biosensors composed of two fusion proteins: a DNA-binding domain fused to the N-terminal portion of Fluc, and a second DNA-binding domain fused to the Fluc C-terminus (Badran et al. 2011). To assess levels of global DNA methylation, both fusion proteins are constructed using the DNA-binding domain of a methyl-CpG-binding domain protein such as MBD2, which has a 100-fold preference for methylated CpG sites over unmethylated CpG sites. The fusion proteins are expressed in a cell-free expression system, then incubated with the target DNA to allow DNA binding. If multiple methylated CpG sites exist in proximity, the N-terminal and C-terminal portions of Fluc will interact (Figure 2). The level of restored Fluc activity is measured using a firefly luciferase assay, such as the Steady-Glo® or Dual-Glo® Luciferase Assay System, and luminescence levels are indicative of DNA methylation levels throughout the genome. To measure site-specific DNA methylation levels, the N-terminus of Fluc is coupled to the MBD DNA-binding domain, but the C-terminus is coupled to a sequence-specific DNA-binding domain (Porter et al. 2008).
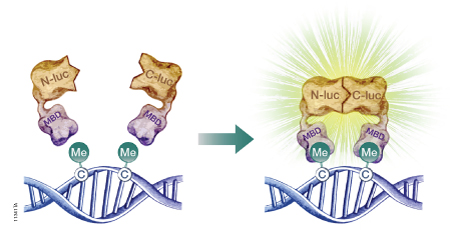
Figure 2. A schematic diagram showing the use of a split-luciferase biosensor to assess DNA methylation at the genome level. To assess DNA methylation at a specific DNA sequence, one of the methyl-CpG-binding domains (MBD) is replaced with a sequence-specific DNA-binding domain. N-luc is the N-terminal portion of firefly luciferase; C-luc is the C-terminal portion of firefly luciferase
Histone Modification and Histone Variants
Epigenetic gene regulation also is controlled by changes in histones that make up the nucleosome and histone modification. Canonical nucleosomes are octamers that consist of H2A, H2B, H3 and H4 proteins. However, there are several histone variants that can vary by a small number of amino acids or include large insertions (reviewed in Sarma and Reinberg, 2005). Often these histone variants are found at specific locations within the chromatin or are used to demarcate boundaries between heterochromatin and euchromatin regions.
The majority of histone-mediated regulation stems from histone modification, most often modification of the exposed amino termini of histones protruding from the nucleosome core. The predominant histone modifications include acetylation, methylation, phosphorylation, ubiquitination and sumoylation, with thousands of potential combinations of modifications within a single nucleosome. Of these, histone acetylation and methylation are the best understood, and some general trends have been observed. Trimethylation of histone H3, specifically the lysine at position 4 (H3K4me3), is a mark associated with transcriptionally active chromatin, whereas H3K27me3 leads to compact chromatin, which represses gene expression. The term “histone code” is used to describe how different combinations of histone modifications affect transcription levels.
Identification of proteins that read, write or erase these marks is critical to help unravel the complexities of epigenetic regulation. Chromatin immunoprecipitation (ChIP) is a powerful assay to identify proteins that bind to chromatin and map protein binding throughout the genome using techniques such as microarray analysis or high-throughput sequencing.
In ChIP analysis, protein:protein and protein:DNA complexes are crosslinked, immunoprecipitated using an antibody against the protein of interest and purified. The DNA sequence of interest then is amplified from the immunoprecipitated material using PCR. Alternatively, the immunoprecipitated DNA can be sequenced (ChIP-seq) or analyzed using microarrays (ChIP-chip) to identify target sequences.

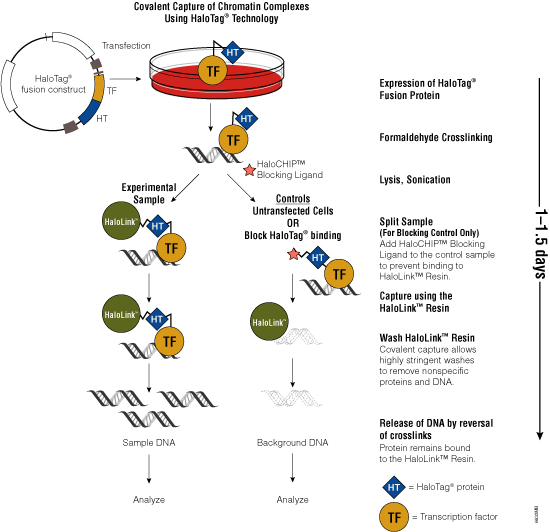
Figure 3. Schematic diagram of the HaloCHIP™ System.

One challenge of the traditional ChIP method is the availability of specific antibodies that recognize crosslinked epitopes. To overcome the need for suitable antibodies, Promega scientists developed the HaloCHIP™ System (Figure 3). This system takes advantage of the HaloTag® protein, which is a mutated hydrolase (Los et al. 2005; Los and Wood, 2007; Los et al. 2008; Hartzell et al. 2009) that catalyzes a covalent attachment to a variety of ligands, including a resin-based ligand for immobilization. This tag can be fused to any protein; for ChIP, the DNA-binding protein of interest is fused to the HaloTag® protein by cloning the protein-coding region into a HaloTag® vector. The recombinant construct is transfected into cells for stable or transient expression, then cells are treated with formaldehyde to induce covalent protein:DNA and protein:protein crosslinks, lysed and sonicated to shear the DNA into smaller fragments. The crosslinked complexes are captured directly from the lysate through covalent binding of the HaloTag® moiety to the HaloLink™ Resin. Covalent binding allows more extensive and stringent washing than is possible with noncovalent interactions, resulting in reduced background and increased signal-to-noise ratio. Subsequent heating of the purified complexes reverses the crosslinks and releases captured DNA fragments, which can be purified and analyzed using PCR, sequencing or microarray analysis. For more information, see the HaloCHIP™ System Technical Manual #TM075.
Histone Acetylation and Deacetylation
Acetylation of a lysine residue neutralizes a positive charge on a histone protein, reducing the electrostatic interaction with negatively charged DNA. This reduction in affinity leads to increased accessibility of the DNA to protein complexes, which can lead to increased gene expression. In addition, lysine acetylation can recruit nucleosome-remodeling complexes, such as Swi2/Snf2, via their bromodomains to promote and maintain euchromatin structure (reviewed in Bernstein et al. 2007). However, the factors controlling gene expression are complex, and histone acetylation also can lead to reduced gene expression through indirect mechanisms.
Lysine acetylation occurs on the N-terminal tails of core histones and is controlled primarily by two enzyme families: histone acetyl transferases (HATs) and histone deactylases (HDACs). HATs use acetyl CoA as a coenzyme to transfer an acetyl group to the epsilon amino group of the lysine side chain. These enzymes are grouped into three families: GNAT, p300/CBP and MYST. HDACs reverse histone acetylation and promote gene silencing. HDACs are often components of large protein complexes and are recruited to sites of DNA methylation by methyl DNA-binding proteins. HDACs fall into four categories: Class I, which includes HDAC1, 2, 3 and 8; Class, II, which includes HDAC4, 5, 6, 7, 9 and 10; Class III, which includes the NAD+-dependent sirtuins (SIRTs); and Class IV, which includes HDAC11 (reviewed in Sun et al. 2012).
Misregulation of HATs and HDACs often is associated with development and progression of cancer and other diseases such as neurodegenerative disorders and cardiovascular diseases, making these enzymes attractive therapeutic drug targets. Many HDAC inhibitors promote cell cycle arrest at the G1/S phase, and studies have shown that tumor cells generally are more sensitive to HDAC inhibitors than normal cells (Johnstone, 2002). Also, HDAC inhibitors can restore the ability of animals to recall memory that had been lost in Alzheimer’s and Parkinson’s disease models, possibly by changing chromatin structure in neurons (Fischer et al. 2007).
To facilitate screening of potential HDAC inhibitors, Promega offers the HDAC-Glo™ I/II Assays and Screening Systems and SIRT-Glo™ Assays Systems. The HDAC-Glo™ I/II and SIRT-Glo™ Assays are single-reagent-addition, homogeneous, luminescent assays that measure relative activities of HDAC class I and II enzymes and sirtuins, respectively. The HDAC-Glo™ I/II Assays use an acetylated, live-cell-permeant, luminogenic peptide substrate that is deacetylated by HDAC activities from cells, extracts or purified enzyme sources (Figure 4). The SIRT-Glo™ Assay uses a similar substrate to detect SIRT activities from purified enzyme sources (Figure 5). Deacetylation of the peptide substrate is measured using a coupled enzymatic system in which a protease in the Developer Reagent cleaves the deacetylated peptide from aminoluciferin, which is quantified in a luciferase-based reaction. The HDAC-mediated luminescent signal is proportional to enzyme activity and persistent, allowing batch processing of multiwell plates in high-throughput screening.
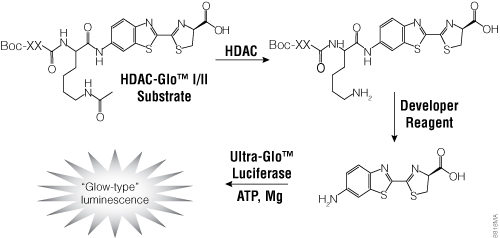
Figure 4. The single-reagent-addition HDAC-Glo™ I/II Assay. HDAC activity deacetylates the luminogenic HDAC-Glo™ I/II Substrate, making the peptide sensitive to specific proteolytic cleavage that is mediated by the HDAC-Glo™ I/II Developer Reagent and liberates aminoluciferin. Free aminoluciferin then can be measured using Ultra-Glo™ firefly luciferase to produce stable, persistent light emission. Boc represents an amino-terminal blocking group that protects the substrate from nonspecific cleavage. XXLysine is an HDAC I/II-optimized consensus sequence derived from histone 4 (Smith et al. 2000).
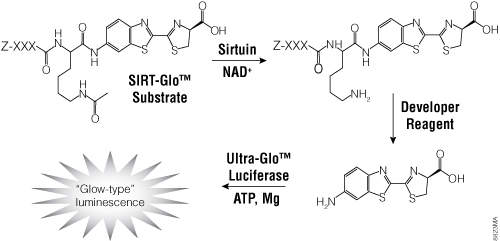
Figure 5. The single-reagent-addition SIRT-Glo™ Assay. SIRT activity deacetylates the luminogenic SIRT-Glo™ Substrate, making the peptide sensitive to specific proteolytic cleavage that is mediated by the SIRT-Glo™ Developer Reagent and liberates aminoluciferin. Free aminoluciferin then can be measured using Ultra-Glo™ firefly luciferase to produce stable, persistent light emission. Z represents an amino-terminal blocking group that protects the substrate from nonspecific cleavage. XXXLysine is a SIRT-optimized amino acid sequence based on a consensus sequence derived from p53 (Abraham et al. 2000).
Additional Resources for the HDAC-Glo™ I/II Assays and Screening Systems
Technical Bulletins and Manuals
TM335 HDAC-Glo™ I/II Assay and Screening System Technical Manual
Histone Methylation
Histone methylation occurs at lysine residues, which can be mono-, di- or trimethylated, and arginine residues, which can be mono- or dimethylated. Histone methylation is catalyzed by protein lysine methyltransferases (PKMTs) and protein arginine methyltransferases (PRMTs) but can be reversed by protein demethylases. To date, researchers have identified >30 demethylating enzymes, >50 protein lysine methyltransferases and >10 protein arginine methyltransferases, suggesting that protein methylation is a dynamic and complex process (Janzen et al. 2010). Histone methylation has different effects on transcriptional activity, depending on the number of methyl groups and position of the amino acid being modified. In general, the H3K9me1 mark is activating, whereas H3K9me2 and H3K9me3 are repressive; H3K4me3 and H3K36me3 are associated with active chromatin, whereas H3K9me3, H3K27me3, H3K36me2 and H4K20me1 often are found in transcriptionally repressed heterochromatin.
The downstream effects of histone methylation are largely determined by proteins that bind to the modified histones. For example, H3K9me3 acts as a binding site for heterochromatin protein 1 (HP1), which then can recruit histone methyltransferases, histone deacetylases and other proteins that affect chromatin structure. H3K4me3 recruits proteins that promote euchromatin, whereas H3K9me1, H3K9me2 and H3K27me3 interact with proteins that promote heterochromatin. Two such groups of proteins are the polycomb group (PcG) proteins and their antagonists, the trithorax (trxG) group proteins, which were first identified as regulators of hox gene expression in Drosophila (Schwartz and Pirrotta, 2008). More recent studies have shown that related proteins exist in mammals and plants. PcG proteins repress transcription; trxG proteins activate transcription. Some PcG and trxG proteins possess histone methyltransferase activity and can modify histones directly, while others bind to and interpret histone modifications.
In embryonic stem (ES) cells, CpG islands that are regulated by PcG proteins often are “bivalent” in that they retain the permissive H3K36me2-depleted and H3K4me3-enriched environment but also exhibit H3K27me3. Genes with bivalent promoters often are actively silenced in ES cells but lose the repressive H3K27me3 mark while retaining the activating H3K4me3 mark later during differentiation.
Histone Phosphorylation
Histones can be phosphorylated on serine, threonine and tyrosine residues. Many of the serine and threonine phosphorylation events play a role in DNA repair or DNA condensation, segregation and decondensation during mitosis, but some are involved in epigenetic regulation of transcription, including H3T3ph, H3T6ph, H3T11ph, H2.AS1ph, H3S10ph and H4S41ph (reviewed in Pérez-Cadahía et al. 2010). H3S10ph is one of the best characterized of these histone modifications. In addition to its DNA-restructuring responsibilities during mitosis, H3S10ph seems important for chromatin decondensation associated with transcriptional activation of target genes. H3S10ph recruits chromatin-modifying enzymes and chromatin-remodeling complexes and prevents binding of HP1 to neighboring H3K9me3 marks at the onset of mitosis. H3S10ph, as well as H3T3ph and H3T11ph, can block binding of DNMT3a to H3, reducing methylation of nearby chromatin (Zhang et al. 2010).
Several kinases are involved in phosphorylation of H3S10, including IkB kinase a (IKKa) (Yamamoto et al. 2003; Anest et al. 2003), proviral integration site for Moloney murine leukemia virus 1 (PIM1) (Zippo et al. 2007) and ribosomal S6 kinase 2 (RSK2) (Sassone-Corsi et al. 1999). Addition of the H3S10ph mark to H3K9me3 is catalyzed by Aurora B kinase (Sabbattini et al. 2007), which also modulates chromosome structure during mitosis and mediates chromosome alignment and attachment to microtubules of the mitotic spindle.
Histones contain many highly conserved tyrosine residues, many of which can be phosphorylated. Phosphorylation of H3Y99 is critical for polyubiquitination and subsequent proteolysis of excess histones, which can increase a cell’s sensitivity to DNA-damaging agents, cause genomic instability and induce apoptosis. Another tyrosine residue, H3Y41, is important in chromatin structure and oncogenesis. In human hematopoietic cell lines, phosphorylation of H3Y41 by Janus kinase 2 (JAK2) destabilizes binding of HP1a to histone H3 (Dawson et al. 2009), leading to a more open chromatin structure around certain gene promoters such as leukemia oncogene LMO2, which can trigger oncogenesis in hematopoietic cells. Overexpression or aberrant activation of JAK2 activity leads to higher levels of H3Y41, loss of HP1a binding and higher expression of LMO2.
Promega offers a number of kinase enzyme systems to monitor the activity or identify inhibitors of different kinases involved in histone phosphorylation, including IKKa, PIM1, RSK2 and several cyclin-dependent kinases (CDKs) such as CDK1, CDK2 and CDK5. These luminescent assays convert ADP produced by these kinases to ATP, which is then converted to light by Ultra-Glo™ Luciferase. The resulting luminescent signal positively correlates with ADP amount and kinase activity. An example protocol is provided below. For a list of available Kinase Enzyme Systems, refer to the Human Kinome chart.
Additional Resources for Histone Phosphorylation
Technical Bulletins and Manuals
TM313 ADP-Glo™ Kinase Assay Technical Manual
Histone Ubiquitination
Conjugation of ubiquitin, a 76-amino acid protein, to lysine residues of histone proteins can affect transcription activity as well as nucleosome stability and, as a result, gene accessibility. The consequences of histone ubiquitination depend on the histone substrate and degree of ubiquitination (reviewed by Weake and Workman, 2008). Mono-ubiquitination of histone H2A (H2Aub1) is often considered a repressive mark, while H2B mono-ubiquitination can play a role in both transcriptional activation and silencing. In addition, there is evidence of cross-talk between histone ubiquitination and other forms of histone modification. For example, ubiquitinated H2B has been identified as a docking site for the COMPASS protein complex (Chandrasekharan et al. 2010), which includes the histone methyltransferase responsible for H3K4 methylation. Also, H2Aub, but not H2A, specifically represses di- and trimethylation of H3K4, and ubiquitin-specific protease 21 (USP21) relieves this repression (Nakagawa et al. 2008).
Ubiquitination of histones can be reversed by cleaving the peptide bond between ubiquitin and the ubiquitinated protein. Several deubiquitinases (DUBs) have been reported to deubiquitylate histones 2A, 2A.Z and 2B, including USP3, USP10, USP21, USP22 and Bap1. Histone deubiquitination has been associated with both transcription activation (Nakagawa et al. 2008; Draker et al. 2011; Gutiérrez et al. 2012) and repression (van der Knaap et al. 2005; van der Knaap et al. 2010).
Sumoylation as a Mechanism of Epigenetic Regulation
Another post-translational modification that plays an important role in epigenetic regulation is sumoylation, the addition of the small ubiquitin-related modifier SUMO (reviewed in Ouyang and Gill, 2009). This modification can stabilize proteins, alter subcellular localization, affect enzyme activity and mediate interactions with other proteins. Many transcription factors and cofactors can be sumoylated, which is generally indicative of transcription repression. In Drosophila, the sumoylated form of Sp3 recruits the polycomb protein Sfmbt (Steilow et al. 2008a) and HP1α, β and γ (Steilow et al. 2008b; Seeler et al. 2001) to repress transcription.
Many histone-modifying enzymes, nucleosome-remodeling complexes and their associated enzyme cofactors contain one or more SUMO interaction motifs (SIMs). This motif allows these proteins to interact with sumoylated transcription factors and cofactors, which can direct these enzymes to specific promoters. Two such groups of histone-modifying enzymes recruited by SUMO are histone deacetylases, which decrease histone acetylation at the target promoter, and histone demethylases such as lysine-specific demethylase 1, which catalyzes the removal of methyl groups from H3K4.
Interpreting DNA Methylation and the Histone Code
Many important regulatory proteins contain domains that bind to modified residues, including plant homeobox domain (PHD) fingers, bromodomains, chromatin organization modifier (chromo) domains, WD40 repeat and tudor domains. Transcription-friendly H3K4me3 acts as a binding site for effector proteins that contain a PHD finger, such as nucleosome remodeling factor (NURF) and the ING4-containing histone acetyltransferase complex. The H3K36me2 modification, which interferes with transcription initiation, acts as a binding site for the chromodomain of the RPD3S histone deacetylase complex.
Histone modifications also can act in cooperation. The specific combination of histone modifications at a particular site often determines which protein complexes and accessory proteins are recruited to activate or repress transcription directly, catalyze additional histone modifications or recruit other histone-modifying proteins. This cooperativity can be explained, at least in part, by the fact that these proteins can contain one or more modified-histone-binding domains. For example, the TFIID protein complex contains both a PHD finger and bromodomain and so binds more strongly to H3K4me3 marks near acetylated H3K9 and H3K14 residues.
The absence of one of these domains through gene mutation or rearrangement can cause serious lapses in gene regulation and diseases such as cancer. Recently, researchers characterized a translocation involving histone demethylase KDM5A that resulted in fusion of the H3K4me3-binding PHD finger of KDM5A to the transcriptional activator NUP98, a common leukemia translocation partner, in an acute myeloid leukemia patient (Islam et al. 2011). Similarly, mixed lineage leukemia (MLL) family members, which act as histone methyltransferases, are involved in translocations in MLL.
Noncoding RNAs
There is increasing evidence that expression of noncoding RNAs, such as microRNAs, small RNAs and large RNAs, play a role in epigenetic gene regulation (reviewed in Costa, 2008; Chuang and Jones, 2007). Noncoding RNAs can direct both cytosine methylation and histone modification to silence DNA repeats in the genome. For example, a class of 29-nucleotide RNAs that was first discovered through their interaction with the spermatogenesis-specific PIWI protein (piwi-interacting RNAs; piRNAs) map to repetitive DNA sequences and are important for silencing short interspersed elements (SINEs), long interspersed elements (LINEs) and long terminal repeat (LTR) retrotransposons.
Several noncoding RNAs also are implicated in X chromosome inactivation. The 17kb X-inactive specific transcript (XIST) RNA binds and coats the inactive X chromosome and forms complexes that modify chromatin structure to suppress transcription. XIST levels are controlled by another large noncoding RNA, TSIX, which is transcribed from the strand opposite of XIST. Noncoding RNAs are involved in genomic imprinting via a similar mechanism (reviewed by Koerner et al. 2009).
Epigenetic Inheritance
Maintenance and inheritance of epigenetic marks during cell division is critical to maintain a committed cell lineage and cellular phenotype in progeny cells, and set a memory of transcriptional status. The transmission of epigenetic information through multiple cell divisions involves many of the mechanisms discussed in this chapter: DNA methylation, histone modification, histone variants and expression of noncoding RNAs (reviewed in Zaidi et al. 2011). These same mechanisms govern the inheritance of epimutations, which can lead to changes in chromatin structure and transcription levels of genes important to diseases such as cancer and imprinting disorders.
Epigenetics and Disease
Aberrant regulation of epigenetic mechanisms can result in genomic imprinting disorders, such as Angelman syndrome and Prader-Willi syndrome, and may contribute to the heritability of many forms of cancer, asthma, Alzheimer’s disease and autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis and multiple sclerosis (reviewed in Hirst and Marra, 2009; Hewagama and Richardson, 2009; Handel et al. 2010). Epimutations can interfere with epigenetic regulation at many levels, including DNA methylation, histone modification and noncoding RNAs. Some epimutations are inherited, but many accumulate due to environmental factors or with age. For example, even though monozygotic twins are epigenetically indistinguishable at birth, their patterns of DNA methylation and histone acetylation can differ dramatically as they age (Fraga et al. 2005).
Related Products
References
- Abraham, J. et al. (2000) Post-translational modification of p53 protein in response to ionizing radiation analyzed by mass spectrometry. J. Mol. Biol. 295, 853–64.
- Anest, V. et al. (2003) A nucleosomal function for IκB kinase-a in NF-κB-dependent gene expression. Nature 423, 659–63.
- Badran, A.H. et al. (2011) Evaluating the global CpG methylation status of native DNA utilizing a bipartite split-luciferase sensor. Anal. Chem. 83, 7151–7.
- Bernstein, B.E., Meissner, A. and Lander, E.S. (2007) The mammalian epigenome. Cell 128, 669–81.
- Bestor, T.H., Hellewell, S.B. and Ingram, V.M. (1984) Differentiation of two mouse cell lines is associated with hypomethylation of their genomes. Mol. Cell. Biol. 4, 1800–6.
- Bianco, T., Hussey, D. and Dobrovic, A. (1999) Methylation-sensitive, single-strand conformation analysis (MS-SSCA): A rapid method to screen for and analyze methylation. Hum. Mutat. 14, 289–93.
- Bird, A.P. (1980) DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 8, 1499–504.
- Bird, A.P. (1986) CpG-rich islands and the function of DNA methylation. Nature 321, 209–13.
- Blackledge, N.P. and Klose, R.J. (2011) CpG island chromatin: A platform for gene regulation. Epigenetics 6, 147–52.
- Carey, N., Marques, J. and Reik, W. (2011) DNA demethylases: A new epigenetic frontier in drug discovery. Drug Discov. Today 16, 683–90.
- Chandrasekharan, M.B., Huang, F. and Sun, Z.W. (2010) Histone H2B ubiquitination and beyond: Regulation of nucleosome stability, chromatin dynamics and the trans-histone H3 methylation. Epigenetics 5, 460–8.
- Chuang, J.C. and Jones, P.A. (2007) Epigenetics and microRNAs. Pediatr. Res. 61, 24R–29R.
- Costa, F.F. (2008) Non-coding RNAs, epigenetics and complexity. Gene 410, 9–17.
- Dawson, M.A. et al. (2009) JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 461, 819–22.
- Deaton, A.M. and Bird, A. (2011) CpG islands and the regulation of transcription. Genes Dev. 25, 1010–22.
- Draker, R., Sarcinella, E. and Cheung, P. (2011) USP10 deubiquitylates the histone variant H2A.Z and both are required for androgen receptor-mediated gene activation. Nucleic Acids Res. 39, 3529–42.
- Farthing, C.R. et al. (2008) Global mapping of DNA methylation in mouse promoters reveals epigenetic reprogramming of pluripotency genes. PLoS Genet. 4, e1000116.
- Fischer, A. et al. (2007) Recovery of learning and memory is associated with chromatin remodelling. Nature 447, 178–82.
- Fraga, M.F. et al. (2005) Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 102, 10604–9.
- Frommer, M. et al. (1992) A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 89, 1827–31.
- Gutiérrez, L. et al. (2012) The role of the histone H2A ubiquitinase Sce in Polycomb repression. Development 139, 117–27.
- Handel, A.E. et al. (2010) Epigenetics: Molecular mechanisms and implications for disease. Trends Mol. Med. 16, 7–16.
- Hartzell, D.D. et al. (2009) A functional analysis of the CREB signaling pathway using HaloCHIP-chip and high throughput reporter assays. BMC Genomics 10, 497.
- Hewagama, A. and Richardson, B. (2009) The genetics and epigenetics of autoimmune diseases. J. Autoimmunity 33, 3–11.
- Hirst, M. and Marra, M.A. (2009) Epigenetics and human disease. Int. J. Biochem. Cell Biol. 41, 136–46.
- Islam, A.B. et al. (2011) Selective targeting of histone methylation. Cell Cycle 10, 413–24.
- Ito, S. et al. (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–3.
- Janzen, W.P. et al. (2010) Epigenetics: Tools and technologies. Drug Discov. Today Technol. 7, e59–e65.
- Johnstone, R.W. (2002) Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 1, 287–99.
- Koerner, M.V. et al. (2009) The function of non-coding RNAs in genomic imprinting. Development 136, 1771–83.
- Kriaucionis, S. and Heintz, N. (2009) The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–30.
- Los, G.V. and Wood, K. (2007) The HaloTag: A novel technology for cell imaging and protein analysis. Methods Mol. Biol. 356, 195–208.
- Los, G.V. et al. (2005) HaloTag® interchangeable labeling technology for cell imaging and protein capture. Cell Notes 11, 2–6.
- Los, G.V. et al. (2008) HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 3, 373–82.
- Meissner, A. et al. (2008) Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–70.
- Mohn, F. et al. (2008) Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 30, 755–66.
- Nakagawa, T. et al. (2008) Deubiquitylation of histone H2A activates transcriptional initiation via trans-histone cross-talk with H3K4 di- and trimethylation. Genes Dev. 22, 37–49.
- Okano, M. et al. (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–57.
- Ouyang, J. and Gill, G. (2009) SUMO engages multiple corepressors to regulate chromatin structure and transcription. Epigenetics 4, 440–4.
- Pérez-Cadahía, B. et al. (2010) Current understanding and importance of histone phosphorylation in regulating chromatin biology. Curr. Opin. Drug Discov. Devel. 13, 613–22.
- Pfaffeneder, T. et al. (2011) The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew. Chem. Int. Ed. Engl. 50, 7008–12.
- Porter, J.R. et al. (2008) A general and rapid cell-free approach for the interrogation of protein-protein, protein-DNA, and protein-RNA interactions and their antagonists utilizing split-protein reporters. J. Am. Chem. Soc. 130, 6488–97.
- Pradhan, S. et al. (1999) Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J. Biol. Chem. 274, 33002–10.
- Reinders, J. et al. (2008) Genome-wide, high-resolution DNA methylation profiling using bisulfite-mediated cytosine conversion. Genome Research 18, 469–76.
- Sabbattini, P. et al. (2007) A novel role for the Aurora B kinase in epigenetic marking of silent chromatin in differentiated postmitotic cells. EMBO J. 26, 4657–69.
- Sarma, K. and Reinberg, D. (2005) Histone variants meet their match. Nat. Rev. Mol. Cell Biol. 6, 139–49.
- Sassone-Corsi, P. et al. (1999) Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science 285, 886–91.
- Schatz, P. et al. (2006) Novel method for high throughput DNA methylation marker evaluation using PNA-probe library hybridization and MALDI-TOF detection. Nucleic Acids Res. 34, e59.
- Schwartz, Y.B. and Pirrotta, V. (2008) Polycomb complexes and epigenetic states. Curr. Opin. Cell Biol. 20, 266–73.
- Seeler, J.S. et al. (2001) Common properties of nuclear body protein SP100 and TIF1alpha chromatin factor: Role of SUMO modification. Mol. Cell Biol. 21, 3314–24.
- Smith, E.R. et al. (2000) The Drosophila MSL complex acetylates histone H4 at lysine 16, a chromatin modification linked to dosage compensation. Mol. Cell. Biol. 20, 312–8.
- Stielow, B. et al. (2008a) Identification of SUMO-dependent chromatin-associated transcriptional repression components by a genome-wide RNAi screen. Mol. Cell 29, 742–54.
- Stielow, B. et al. (2008b) SUMO-modified Sp3 represses transcription by provoking local heterochromatic gene silencing. EMBO Rep. 9, 899–906.
- Sun, W.J. et al. (2012) Histone acetyltransferases and deacetylases: Molecular and clinical implications to gastrointestinal carcinogenesis. Acta Biochim. Biophys. Sin. (Shanghai) 44, 80–91.
- van der Knaap, J.A. et al. (2005) GMP synthetase stimulates histone H2B deubiquitylation by the epigenetic silencer USP7. Mol. Cell 17, 695–707.
- van der Knaap, J.A. et al. (2010) Biosynthetic enzyme GMP synthetase cooperates with ubiquitin-specific protease 7 in transcriptional regulation of ecdysteroid target genes. Mol. Cell. Biol. 30, 736–44.
- Weake, V.M. and Workman, J.L. (2008) Histone ubiquitination: Triggering gene activity. Mol. Cell 29, 653–63.
- Weber, M. et al. (2007) Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 39, 457–66.
- Wojdacz, T.K. and Dobrovic, A. (2007) Methylation-sensitive high resolution melting (MS-HRM): A new approach for sensitive and high-throughput assessment of methylation. Nucleic Acids Res. 35, e41.
- Yamamoto, Y. et al. (2003) Histone H3 phosphorylation by IKK-a is critical for cytokine-induced gene expression. Nature 423, 655–9.
- Zaidi, S.K. et al. (2011) Bookmarking the genome: Maintenance of epigenetic information. J. Biol. Chem. 286, 18355–61.
- Zhang, X. et al. (2006) Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 126, 1189–201.
- Zhang, Y. et al. (2010) Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 38, 4246–53.
- Zippo, A. et al. (2007) PIM1-dependent phosphorylation of histone H3 at serine 10 is required for MYC-dependent transcriptional activation and oncogenic transformation. Nat. Cell Biol. 9, 932–44.
Dual-Glo, Flexi, HaloTag, Nano-Glo, Steady-Glo and Wizard are registered trademarks of Promega Corporation. ADP-Glo, Find My Gene, HaloCHIP, HaloLink, HDAC-Glo, MethylEdge, SIRT-Glo and Ultra-Glo are trademarks of Promega Corporation.